
Reactive Oxygen Species Regulate Caspase-11 Expression and Activation of the Non-canonical NLRP3 Inflammasome during Enteric Pathogen Infection
Caspase-11 is required for NLRP3 inflammasome activation and cell death in response to certain gram-negative bacterial infections like Citrobacter rodentium. However, how C. rodentium drives caspase-11 expression and activation is not well understood. Here, we demonstrate that the NOD2-RIP2 pathway regulates reactive oxygen species production and c-Jun N-terminal kinase signaling to control caspase-11 expression and subsequent activation of caspase-11 and the NLRP3 inflammasome during C. rodentium infection. In the absence of NOD2-RIP2 signaling, increased inflammasome activation results in lower bacteria numbers in the colon and less tissue damage during the early stages of infection.
Published in the journal:
Reactive Oxygen Species Regulate Caspase-11 Expression and Activation of the Non-canonical NLRP3 Inflammasome during Enteric Pathogen Infection. PLoS Pathog 10(9): e32767. doi:10.1371/journal.ppat.1004410
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004410
Summary
Caspase-11 is required for NLRP3 inflammasome activation and cell death in response to certain gram-negative bacterial infections like Citrobacter rodentium. However, how C. rodentium drives caspase-11 expression and activation is not well understood. Here, we demonstrate that the NOD2-RIP2 pathway regulates reactive oxygen species production and c-Jun N-terminal kinase signaling to control caspase-11 expression and subsequent activation of caspase-11 and the NLRP3 inflammasome during C. rodentium infection. In the absence of NOD2-RIP2 signaling, increased inflammasome activation results in lower bacteria numbers in the colon and less tissue damage during the early stages of infection.
Introduction
Enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) infections in humans are a major source of morbidity and mortality, especially in developing countries [1]. To study these infections, Citrobacter rodentium is used as an enteric bacterial pathogen of mice that triggers similar inflammatory responses to those observed in humans infected with EPEC and EHEC [2]. As with EPEC and EHEC in humans, C. rodentium infection in the mouse results in bacterial attachment and effacing lesion formation in the lumen of the colon [3]. Clearance of C. rodentium from the host tissues requires contributions from humoral and Th1 immune responses [4]–[6]. However, innate immunity is also important for early bacterial control [7], [8] and for the initiation of adaptive immunity [9], [10].
Innate immunity to pathogens depends on a limited set of germ-line encoded pattern recognition receptors (PRRs) that sense conserved pathogen motifs. Innate receptors initiate inflammation through activation of pro-inflammatory transcription factors such as NF-κB, by promoting activation of proinflammatory caspases in inflammasomes, and by initiating programmed cell death of infected cells. The intracellular NOD-like receptors (NLRs) NOD1 and NOD2 recognize peptidoglycan fragments from bacterial cell walls in the cytosol, which results in pro-inflammatory NF-κB and MAP-kinase pathway activation via the adaptor RIP2 [11]–[13]. However, pathogen infection is frequently accompanied by ion fluxes and vacuolar membrane damage elicited by the action of microbial toxins and effectors of specialized bacterial secretion systems [14]–[17]. Thus, while conserved bacterial peptidoglycan fragments are initially recognized by receptors NOD1 and NOD2, the perturbations inflicted over the course of infection can activate NLRP3.
NLRP3 is the most studied inflammasome-associated NLR. Signaling events initiated by Toll-like receptors (TLRs) contribute to NF-κB mediated up-regulation of NLRP3 and pro-IL-1β for robust activation [18]. No specific pathogen-derived ligand is known to bind and activate NLRP3. Instead, NLRP3 is activated in response to diverse stimuli including microbial, environmental and metabolic perturbations, which culminate in the generation of reactive oxygen species, changes in ion flux or leakage of cathepsin B into the cytoplasm [19]–[21]. These cellular damage signals induce NLRP3 to form an inflammasome complex with the adaptor ASC and the cysteine protease caspase-1 that enables the maturation of IL-1β and IL-18 cytokines and pyroptotic cell death [22]–[25]. An alternative non-canonical pathway of NLRP3 activation that additionally involves upstream caspase-11 activation has been described in response to cytosolic LPS and gram-negative enteric pathogens such as Citrobacter rodentium and Escherichia coli [26], [27]. Recent studies have extended the requirement of the non-canonical inflammasome to other gram-negative pathogens such as flagellin-deficient Legionella pneumophila (ΔflaA) [28] and Salmonella typhimurium (Δfljb/flic) [29]. Studies from our lab and others suggest that activation of the non-canonical NLRP3 inflammasome requires TLR4-TRIF mediated type-I interferon dependent production of caspase-11 [30], [31]. However, whether NLR signaling pathways can regulate the non-canonical NLRP3 inflammasome is not known. Here, we show that activation of the cytoplasmic NOD2-RIP2 pathway modulates caspases-11 expression and activation of the non-canonical inflammasome in response to C. rodentium and Δfljb/flic Salmonella typhimurium infection.
Results
Nod2 - and Rip2-deficient cells display enhanced inflammasome activation
Signaling events initiated by TLRs contribute to NF-κB mediated up-regulation of NLRP3 and pro-IL-1β [18]. However, whether TLR or NLR pathways can restrain or negatively regulate inflammasome activation is not known. To examine the role of NOD2 in inflammasome activation, WT and Nod2−/− bone marrow derived macrophages (BMDM) were infected with C. rodentium and caspase-1 activation was analyzed at 18 h post-infection. Nod2-deficient cells exhibited increased caspase-1 activation and this was accompanied with elevated production of IL-18 (Figure 1A–C). Similarly, Rip2−/− cells displayed increased caspase-1 activation and IL-18 secretion (Figure 1D–F). Furthermore, increased caspase-1 activation was not due to increased bacterial loads because Nod2−/− and Rip2−/− macrophages exhibited comparable C. rodentium uptake and clearance (Fig. 1G). These results suggest that the NOD2-RIP2 axis negatively regulates inflammasome activation to C. rodentium infection.
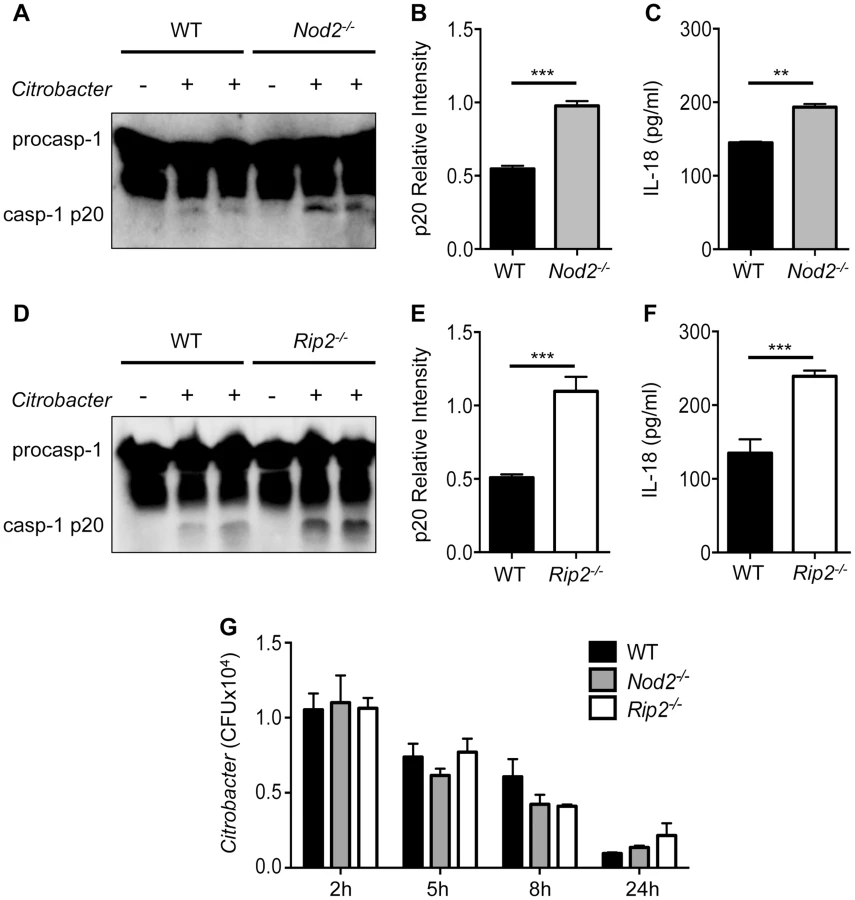
The NOD2-RIP2 axis specifically restricts the NLRP3 inflammasome
Activation of caspase-1 is triggered downstream of several NLRs including NLRP3, NLRP1 and NLRC4 [23], [32], [33]. Previously, we have shown that infection with C. rodentium specifically activates the non-canonical NLRP3 inflammasome [30]. To determine the extent of NOD2-RIP2 modulation, we examined whether this pathway also modulates the NLRC4 inflammasome. To answer this, we infected WT, Nod2 - and Rip2-deficient cells with Salmonella typhimurium to activate the NLRC4-inflammasome. However, comparable caspase-1 activation was observed in S. typhimurium infected WT, Nod2−/− and Rip2−/− cells (Figure 2A,B). In agreement, IL-18 levels were similar (Figure 2C) suggesting that the NOD2-RIP2 pathway specifically restricts the NLRP3 inflammasome. The NLRC4 inflammasome is activated upon recognition of S. typhimurium flagellin [34], [35]. However, flagellin-deficient S. typhimurium Δfljb/flic activates the NLRP3 inflammasome [29]. Consequently, Nod2−/− or Rip2−/− BMDM infected with the S. typhimurium Δfljb/flic strain displayed enhanced caspase-1 activation and IL-18 production over wildtype BMDM (Figure 2D–F). These results suggest that NOD2 and RIP2 specifically modulated the NLRP3 inflammasome in this model while the NLRC4 inflammasome was not affected by the absence of the NOD2-RIP2 signaling axis.
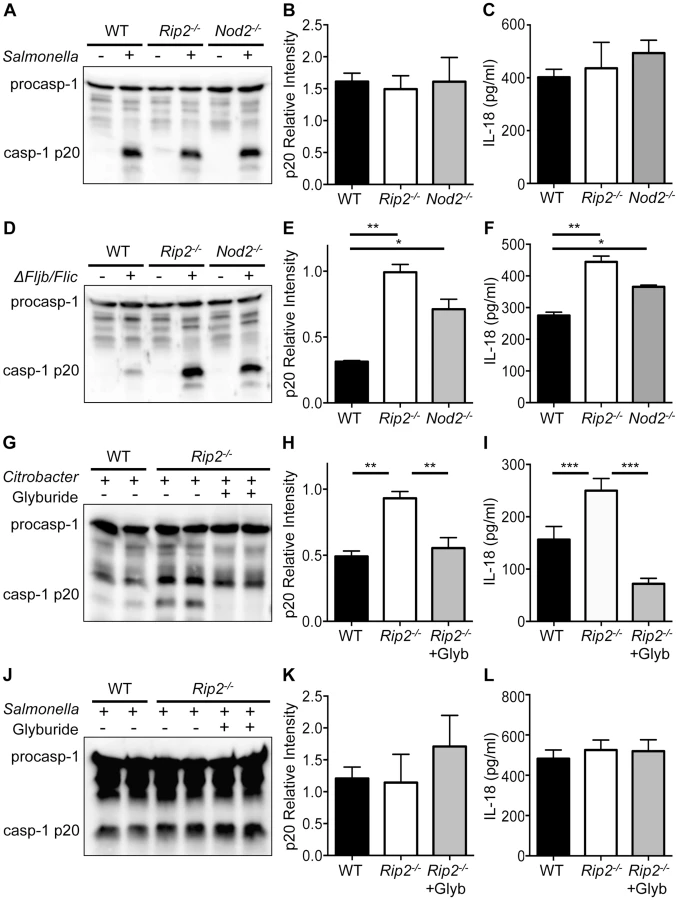
Glyburide, a type 2 diabetes drug, has been shown to specifically inhibit the NLRP3 inflammasome in response to microbial and crystalline stimuli [36]. To further verify that the enhanced caspase-1 activation observed in Nod2 - and Rip2-deficient BMDM is the result of elevated NLRP3 inflammasome activation, we exposed Rip2-deficient BMDM to glyburide following C. rodentium infection. Consistent with the requirement for NLRP3, we observed increased caspase-1 activation in Rip2-deficient cells and this was markedly abrogated in the presence of glyburide (Figure 2G,H). In agreement, enhanced IL-18 levels in Rip2−/− BMDM were significantly decreased upon treatment with glyburide (Figure 2I). In contrast, glyburide treatment did not affect caspase-1 activation in Rip2-deficient cells infected with S. typhimurium (Figure 2J,K). Consequently, IL-18 levels were also unaffected (Figure 2L). These data indicate that NOD2-RIP2 signaling specifically modulates NLRP3 activation and does not regulate all inflammasomes.
Rip2-deficient cells show increased ROS production
To determine the cause of increased NLRP3 inflammasome activation during C. rodentium infection, we examined the possibility that there is a global dysregulation of cytokines and inflammation in Rip2-deficient BMDM. However, there was no difference in IL-6 production and TNF-α levels were lower in Rip2-deficient cells (Figure 3A) suggesting that a global increase in inflammation is not responsible for increased NLRP3 inflammasome activation in this model.

Elevated production of reactive oxygen species (ROS) has previously been associated with increased NLRP3 inflammasome activation [37]. In particular, mitochondrial-derived ROS production was suggested to provoke NLRP3 activation [38], and we previously demonstrated that Rip2-deficient cells have defects in autophagy that lead to the accumulation of damaged mitochondria [39]. LC3 is an autophagy-associated protein that is localized in the cytosol under steady state conditions, but relocalizes to autophagosome membranes when autophagy occurs [40]. During C. rodentium infection of WT or Rip2−/− BMDM, the number of autophagosomes was examined using confocal microscopy by counting GFP-LC3+ puncta per cell (Figure 3B–C). Alternatively, we used flow cytometry to measure the fluorescence intensity of GFP-LC3 after permeablizing cells so that only autophagosome membrane bound GFP-LC3 remained (Figure 3D). In both cases, we found that autophagy was impaired in Rip2−/− BMDM compared to WT cells after C. rodentium infection. Furthermore, Rip2-deficient BMDM exhibited elevated levels of mitochondria-derived superoxide when infected with C. rodentium and stained with the mitochondrial specific ROS sensor MitoSOX (Figure 3E,F). Finally, Rip2−/− BMDM treated with the ROS scavenger N-acetyl-L-cysteine (NAC) had reduced caspase-1 activation and IL-18 production compared to WT cells (Figure 3G–I). Thus, the presence of dysfunctional mitochondria in Rip2−/− cells leads to enhanced ROS production and thereby increased NLRP3 inflammasome activation.
Caspase-11 activation is upregulated in Rip2-deficient cells
Enteric pathogens such as C. rodentium are known to activate the NLRP3 inflammasome by a non-canonical pathway, which additionally requires caspase-11 for its activation [26]. We therefore postulated that during C. rodentium infection increased ROS production might act upstream of caspase-11 as well as caspase-1. Therefore, we sought to examine the role of caspase-11 in the present study. First, we determined that activation of caspase-11 is enhanced in Rip2−/− BMDM (Fig. 4A,B). Next, we treated Rip2−/− BMDM with the ROS scavenger NAC following C. rodentium infection and observed that caspase-11 activation was significantly diminished compared to mock-treated Rip2−/− BMDM controls (Figure 4C,D). We also found that caspase-11 activation (p30 band) correlated with the expression of p43/p38 (pro-caspase-11). Indeed, densitometric analysis showed that C. rodentium infected Rip2−/− BMDM had higher expression of pro-caspase-11 and that treatment with NAC inhibited pro-caspase-11 expression (Figure 4E). Mechanistically, we examined whether the increased caspase-11 seen in Rip2−/− BMDM was the result of defects in protein turnover; however, treatment of WT and Rip2−/− BMDM 4 h after infection with the proteasome inhibitor MG-132 resulted in accumulation of caspase-11 in both WT and Rip2−/− BMDM (Figure 4F–G). Instead, Casp11 mRNA was higher in Rip2−/− BMDM compared to WT BMDM (Figure 4H). These results indicate that RIP2 regulates caspase-11 at the level of mRNA expression, possibly through enhanced ROS production.

Exogenous ROS enhances caspase-11 expression and activation in WT cells
To further confirm the role of enhanced ROS production leading to increased inflammasome activation in Rip2−/− BMDM, we examined the effects of treating WT BMDM with exogenous ROS. WT BMDM were infected with C. rodentium and treated with H2O2 as a ROS source 6 h after infection. Although H2O2 treatment had no effect on Il18 mRNA levels following C. rodentium infection, we detected significantly more IL-18 in the culture medium (Figure 5A,B). In agreement, caspase-1 activation was also increased following H2O2 treatment (Figure 5C). Importantly, H2O2 treatment of WT cells resulted in increased mRNA and protein expression of caspase-11 by qRT-PCR and Western blot respectively (Figure 5D–F). Our results indicate that ROS produced during C. rodentium infection can enhance caspase-11 expression and subsequently potentiate activation of the non-canonical inflammasome.

ROS regulates caspases-11 through a JNK mediated pathway
Our data indicate that ROS is capable of regulating caspase-11 expression. However, it remains unclear what pathways are regulated by ROS that subsequently affect caspase-11 expression. As a TLR4-TRIF-IFN-β pathway has been established for expression of caspase-11 [30], [31], we examined the level of type-I interferons, but found they were similar between WT, Rip2−/−, and Nod2−/− BMDM (Figure 6A). One pathway that is responsive to ROS is the c-Jun N-terminal kinase (JNK) pathway [41]. We next infected WT BMDM with C. rodentium, treated them with H2O2 and examined the effects on JNK1/2 phosphorylation. H2O2 treatment resulted in increased JNK phosphorylation compared to mock treated cells (Figure 6B). Similarly, Rip2−/− BMDM infected with C. rodentium alone had substantially more JNK1/2 phosphorylation than WT cells (Figure 6C). Finally, we infected WT or Rip2−/− BMDM with C. rodentium and then treated Rip2−/− BMDM with the JNK inhibitor SP600125. We observed that caspase-11 levels were reduced in conjunction with reduced JNK1/2 activation (Figure 6D). From these results we propose that in the absence of NOD2-RIP2 signaling, defective autophagy results in the accumulation of ROS and the subsequent enhancement of JNK signaling. This ultimately leads to an increase in caspase-11 expression and non-canonical inflammasome activation (Figure 6E).

Increased non-canonical inflammasome activation provides protection against C. rodentium induced colitis
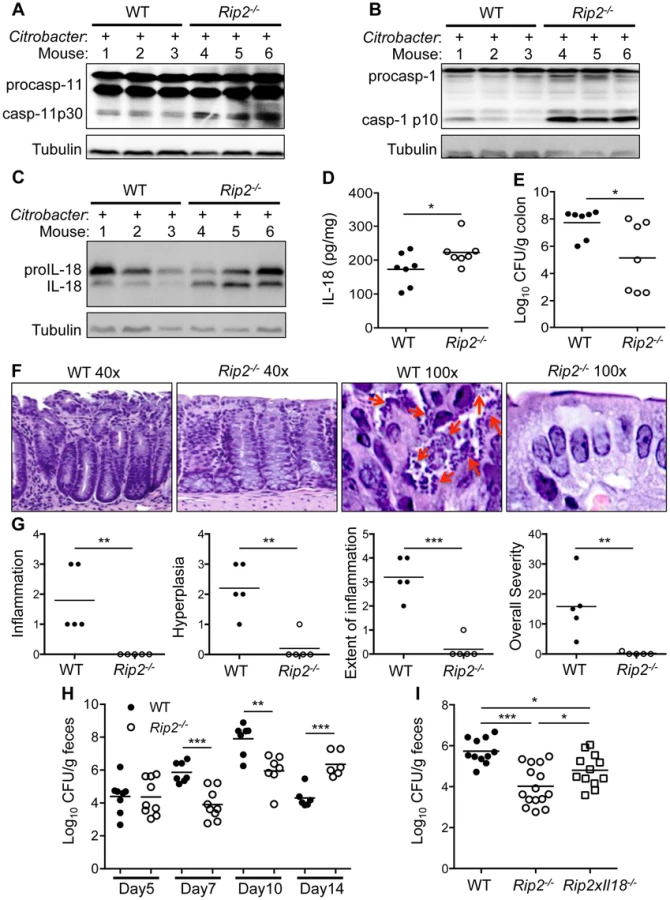
To determine the physiological relevance of our proposed pathway, we examined Rip2-deficient mice for IL-18 production and inflammasome activation in the colon during C. rodentium induced colitis. On day 10 post-infection, caspase-11 and caspase-1 activation were much higher in colon lysates of Rip2−/− mice compared to WT controls (Figure 7A,B). IL-18 activation was significantly increased by Western blot and IL-18 levels were higher in colon lysates taken from Rip2−/− mice compared to WT controls (Figure 7C,D). In contrast, bacterial loads in the colon were reduced in Rip2−/− versus WT mice (Figure 7E). In agreement, colon tissue examined on day 10 after infection showed increased numbers of bacteria adherent to the mucosal surface in WT compared to Rip2−/− mice (Figure 7F). Intriguingly, colon sections from WT mice also showed more hyperplasia and inflammation and the extent of colon area that was inflamed was more pronounced with WT mice compared to Rip2−/− mice (Figure 7F,G). The stool of Rip2−/− mice also had fewer C. rodentium CFU at both day 7 and day 10, highlighting the protective effect of increased inflammasome activation during the innate immune phase of infection (Figure 7H). These results demonstrate that the NOD2-RIP2 pathway plays an important role in regulating the non-canonical NLRP3 inflammasome in vivo during C. rodentium-induced colitis. Of note, previous reports have demonstrated an essential role for adaptive immunity in the eventual clearance of C. rodentium [9], [42]. In agreement with these studies, on day 14 after infection, mice deficient in RIP2 signaling had a clearance defect in the stool (Figure 7H). Finally, to verify that increased inflammasome activation and IL-18 production were responsible for the lower bacterial numbers on day 7 or 10, we infected WT, Rip2−/−, and Rip2−/−×Il18−/− mice with C. rodentium and examined bacterial loads on day 7 after infection. As before, Rip2−/− mice had significantly lower bacterial numbers compared to WT mice. However, the deletion of IL-18 in Rip2−/−×Il18−/− mice resulted in significantly increased bacterial burden compared to the Rip2−/− mice (Figure 7I).
Discussion
Regulation of the non-canonical NLRP3 inflammasome is still not well understood. It is clear that pathogen entry into the cytosol is required for caspase-11 activation [17], [43], [44]. In addition, caspase-11 activation requires guanylate binding protein (GBP) for activation in response to cytosolic LPS [45]. However, the exact upstream LPS sensor has yet to be identified [46], [47]. Although the exact activation mechanism involved is not clear, our lab and others demonstrated that expression of caspase-11 via a TLR4-TRIF-IFN-β-dependent mechanism is necessary for non-canonical NLRP3 inflammasome activation during C. rodentium infection [30], [31]. Our current findings show that ROS produced during C. rodentium infection, or added exogenously to infected cells as H2O2, is capable of regulating caspase-11 expression. In agreement, treatment with the ROS scavenger NAC dampened caspase-11 expression and activation, and subsequently reduced caspase-1 activation through the non-canonical NLRP3 inflammasome. Mechanistically, Rip2−/− cells display enhanced ROS production following C. rodentium infection, and our data define a pathway where ROS acts through JNK signaling to increase caspase-11 expression and the subsequent activation of the non-canonical NLRP3 inflammasome.
Similar to C. rodentium, we found that infection with flagellin-deficient S. typhimurium Δfljb/flic resulted in augmented inflammasome activation in Nod2−/− and Rip2−/− BMDM. Since activation of the NLRC4 inflammasome was comparable between WT and Rip2-deficient BMDM infected with WT S. typhimurium, these data suggest that NOD2-RIP2 mediated regulation of ROS and JNK activation specifically modulates the non-canonical NLRP3 inflammasome and not NLRC4.
In addition to the mechanistic insight provided by our in vitro studies, caspase-1 activation has never been examined in Rip2−/− mice in response to C. rodentium. Our findings demonstrate the NOD2-RIP2 pathway helps suppress the non-canonical NLRP3 inflammasome in vivo during C. rodentium induced colitis. In the absence of RIP2, there is increased caspase-11 and caspase-1 activation and higher levels of processed IL-18. These increases are associated with reduced bacterial burden and protection of the colon epithelium during the innate immune phase of infection (d7–d10). Our findings are thus in line with our previous publications, which show that NLRP3 inflammasome activation and IL-18 production are important for the control of bacterial burden and disease during C. rodentium infection [48]. The importance of IL-18 in the protection seen in this model was also confirmed through the use of Rip2−/−×Il18−/− mice. Although IL-18 deletion in Rip2−/− mice did not entirely abolish the protective phenotype, this is likely due to pyroptosis or other functions of caspase-11, such as lysosome-phagosome fusion [49].
Other reports have examined NOD2 signaling in vivo. However, these reports focused on the effects of NOD2-RIP2 signaling on the adaptive immune response. Intriguingly, similar results to ours were published for the ubiquitin ligase pellino3, which is required for NOD2-RIP2 signaling. At early time points, Peli3−/− mice had reduced bacterial burden similar to our model, but after d14, these mice showed a clearance defect [42]. On the other hand, Nod2−/− mice displayed reduced production of the chemokine CCL2, which ultimately lead to impaired adaptive immunity and impaired clearance of C. rodentium after d14 of infection [9]. Collectively, these findings suggest that increased NLRP3 inflammasome activation provides protection early during the innate immune phase, but the defects in adaptive immunity that are also present in Nod2−/−, Rip2−/−, or Peli3−/− mice ultimately lead to an inability of these mice to resolve the infection.
Although inflammation and cell death are important for bacterial clearance [25], they also increase the likelihood of bacterial dissemination to other tissues and induction of severe inflammatory responses by increased tissue damage and released cellular contents [50]–[52]. Therefore, the sequential and carefully orchestrated activation of inflammatory pathways is essential for pathogen clearance as well as maintaining homeostasis in the host. Initial recognition of specific peptidoglycan fragments from bacterial cell walls by the NOD2-RIP2 pathway, results in pro-inflammatory NF-κB and MAP-kinase activation within minutes of infection [53], [54]. However, if this initial inflammatory burst is inadequate, then subsequent inflammatory pathways, such as the non-canonical NLRP3 inflammasome, are initiated. One common feature of pathogens that activate the non-canonical NLRP3 inflammasome is activation proceeds with slower kinetics than during canonical activation. In all instances, non-canonical NLRP3 inflammasome activation is observed only after a time-period of 12 to 16 h [26], [28], [30], [31], whereas canonical activation requires less than 1 h [21], [22], [55]. Based on our results, we propose that NOD2-RIP2 signaling initially suppresses or delays non-canonical NLRP3 inflammasome activation by preventing or removing mitochondrial damage. However, extended assault from C. rodentium eventually overcomes these mechanisms and leads to ROS generation and activation of the non-canonical NLRP3 inflammasome [20], [39], [56]. In the absence of NOD2 or RIP2, increased mitochondrial damage and ROS production leads to elevated JNK activation, and in turn to augmented non-canonical NLRP3 inflammasome activation. During C. rodentium infection in vivo, this impaired initial bacterial colonization of the colon and provided protection of the epithelium up to days 10 after infection. However, in other colitis models, increased inflammasome activation may result in increased damage to the colon tissue if left unchecked. The NOD2-RIP2 pathway has been associated with Crohn's disease in humans [57] and plays an essential role in several mouse models of colitis [58]–[62]. As Rip2-deficiency leads to enhanced inflammasome activation, it will be of interest to examine caspase-1 activation in other models where inflammation is a key factor and where the NOD2-RIP2 axis is involved.
Materials and Methods
Ethics statement
Experiments were conducted under protocols approved by the St. Jude Children's Research Hospital Committee on Use and Care of Animals (Protocol #482) and were performed in accordance with institutional policies, AAALAC guidelines, the AVMA Guidelines on Euthanasia (CO2 asphyxiation followed by cervical dislocation), NIH regulations (Guide for the Care and Use of Laboratory Animals), and the United States Animal Welfare Act (1966).
Mice
All mice were maintained at SJCRH and were backcrossed for at least 10 generations onto the C57BL/6J (B6) background. Nod2−/−, Rip2−/− and Rip2−/−×Il18−/− mice have been reported previously [39], [63], [64]. WT-GFP-LC3+ and Rip2−/−-GFP-LC3+ mice were generated by crossing WT or Rip2−/− mice with LysM-Cre+ Atg7loxP/loxP GFP-LC3+ mice [39] and selecting progeny that only contained the GFP-LC3 transgene but not LysM-Cre or Atg7loxP/loxP. All mice were housed in the SJCRH animal resource center, which is a specific pathogen free (SPF) and AAALAC accredited facility.
Bacterial infection, caspase-1 activation and IL-18 production
WT, Nod2−/− and Rip2−/− bone marrow derived macrophages (BMDM) were differentiated in complete IMDM containing 10% heat-inactivated FBS and supplemented with M-CSF containing L929 supernatant at 37°C in a humidified atmosphere containing 5% CO2 for 5 days. Citrobacter rodentium (ATCC 51459), Salmonella typhimurium (SL1344), Salmonella typhimurium Δfljb/flic were grown in LB broth overnight with shaking at 37°C. Next day, the bacteria were subcultured to mid-log phase, washed in PBS and BMDM were infected with 20 multiplicity of infection (MOI) of C. rodentium, 1MOI of S. typhimurium, or 10MOI of S. typhimurium Δfljb/flic. Cell lysates combined with supernatants were collected after 18 h of infection and assessed for caspase-1 activation by Western blot using the anti-caspase-1 p20 antibody from Adipogen (AG-20B-0042-C100). Western blots were imaged on a Kodak IMM4000pro and the densitometry performed using the Carestream software provided by the manufacturer. Caspase-1 p20 levels were normalized to the proCaspase-1 for that lane. Supernatants collected at 18 h after infection were analyzed for IL-6, TNF-α, or IL-18 by ELISA according to the manufacturer's instructions (eBiosciences) or for type-I interferon by the RAW-Blue ISG cell line (Invivogen).
C. rodentium growth assay in BMDM
WT, Nod2−/− and Rip2−/− macrophages were grown in 24-well plates and infected with 10 MOI of C. rodentium at 37°C. After 2 h of infection, cells were washed three times with PBS and 10 µg/ml of gentamicin was added to kill extracellular bacteria. Cells were lysed in 0.1% Triton X-100 at indicated times and lysates were serially diluted and plated on LB agar plates. The colonies were counted after 18 h of growth at 37°C
Treatments
For SP600125 (JNK inhibitor, 10 µM, Santa Cruz Biotechnology), N-acetyl-L-cysteine (NAC, 10 mM, Sigma) or glyburide (200 µM, Sigma) treatment, BMDMs were exposed to these inhibitors after 2 h of C. rodentium infection. For H2O2 treatment, BMDM were treated at 6 h after infection. For MG-132 (1 µM) treatment, BMDM were treated at 4 h after infection, at 2, 4, 8 or 18 h after infection, supernatants were collected for ELISA and cells or cells + supernatants were lysed with RIPA buffer containing protease inhibitors and phosphatase inhibitors (Calbiochem), followed by boiling in sodium dodecyl sulfate (SDS) sample buffer and examined by Western blot. Anti-caspase-1 p20 (Adipogen), anti-caspase-1 p10 (Santa Cruz Biotechnology, A20), anti-total JNK (Cell signaling Technologies, 9252), anti-phospho JNK (Cell signaling Technologies, 9251L) and anti-caspase-11 antibodies (Enzo, 4E11) were used for Western blot detection and equal loading was verified by blotting with anti-GAPDH or anti-Tubulin (Cell Signaling Technologies). HRP-labeled anti-mouse or -rabbit antibodies were obtained from Jackson Immuno Research. Caspase-11 densitometry was normalized to the procaspase-11 level in the uninfected lane for each sample. Phospho-JNK densitometry was normalized to GAPDH loading control for each lane. IL-18 levels were determined in the supernatants by ELISA according to manufacturer's instructions (eBioscience). Alternatively, cells were collected at the indicated time points and RNA isolated for analysis by qRT-PCR. Primers: GAPDH forward 5′-CGTCCCGTAGACAAAATGGT-3′ reverse 5′-TTGATGGCAACAATCTCCAC-3′; IL-18 forward 5′-GCCTCAAACCTTCCAAATCA-3′ reverse 5′-TGGATCCATTTCCTCAAAGG-3′; Caspase-11 forward 5′-ACAATGCTGAACGCAGTGAC-3′ reverse 5′-CTGGTTCCTCCATTTCCAGA-3′.
ROS production
For staining mitochondria, uninfected BMDMs were used as controls or infected with 20MOI of C. rodentium for 18 h and then stained for 30 min in fresh medium containing 5 µM MitoSOX. Cells were then washed in HBSS, resuspended in flow cytometry buffer (DPBS with 1% FBS and 0.04% NaN3) and analyzed immediately by flow cytometry.
Autophagy
WT-GFP-LC3+ and Rip2−/−-GFP-LC3+ mice were used to generate BMDM as before and cells were infected with 20MOI of C. rodentium for 4 h or 18 h. Cells were then permeabilized with 0.05% saponin in PBS for 5 min to remove cytosolic GFP-LC3 and cells were fixed in 4% paraformaldehyde in PBS for 5 min. Cells were then examined by fluorescence confocal microscopy for the number of GFP-LC3+ puncta after staining with Alexafluor 647 phalloidin and mounting with ProLong Gold+DAPI (Life Technologies). Alternatively, cell were scraped from wells and resuspended in FACS buffer and examined by flow cytometry for GFP intensity as a readout of autophagy.
Citrobacter rodentium induced colitis
Age and sex matched 8–10 week old mice were infected with 1010 CFU of C. rodentium by oral gavage. Stool was collected from mice for CFU at the indicated time points. Mice were euthanized on D10 and colons collected for CFU determination by homogenizing in PBS or for Western blot by homogenizing in RIPA buffer containing protease inhibitors and phosphatase inhibitors (Calbiochem). Protein concentration was normalized and samples analyzed by western blot; anti-IL-18 (MBL, 39-3F) anti-caspase-1 p10 (Santa Cruz Biotechnology, A20) and anti-caspase-11 antibodies (Enzo, 4E11) were used. Lysates were additionally examined by ELISA for IL-18 (Ebiosciences) and normalized to total protein. D10 colon samples were also collected, formalin fixed and processed for routine histopathological examination by hematoxylin and eosin staining. Sections were examined by a board certified veterinary pathologist (Peter Vogel) for inflammation, bacterial invasion of epithelial surface, and distribution of inflammation.
Statistics
All statistical analyses were performed using GraphPad Prisim 6.0. Students t-Test was used for single comparisons and One-Way ANOVA with Dunnett's or Sidak's post-hoc test for multiple comparisons. Two-way ANOVA was used to analyze C. rodentium growth between groups over time.
Zdroje
1. KaperJB, NataroJP, MobleyHL (2004) Pathogenic Escherichia coli. Nat Rev Microbiol 2 : 123–140.
2. BorenshteinD, McBeeME, SchauerDB (2008) Utility of the Citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol 24 : 32–37.
3. MundyR, MacDonaldTT, DouganG, FrankelG, WilesS (2005) Citrobacter rodentium of mice and man. Cell Microbiol 7 : 1697–1706.
4. KamadaN, KimYG, ShamHP, VallanceBA, PuenteJL, et al. (2012) Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336 : 1325–1329.
5. SimmonsCP, ClareS, Ghaem-MaghamiM, UrenTK, RankinJ, et al. (2003) Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun 71 : 5077–5086.
6. BryL, BrennerMB (2004) Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172 : 433–441.
7. GibsonDL, MaC, BergstromKS, HuangJT, ManC, et al. (2008) MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol 10 : 618–631.
8. GibsonDL, MaC, RosenbergerCM, BergstromKS, ValdezY, et al. (2008) Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol 10 : 388–403.
9. KimYG, KamadaN, ShawMH, WarnerN, ChenGY, et al. (2011) The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34 : 769–780.
10. GeddesK, RubinoSJ, MagalhaesJG, StreutkerC, Le BourhisL, et al. (2011) Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17 : 837–844.
11. GirardinSE, TournebizeR, MavrisM, PageAL, LiX, et al. (2001) CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep 2 : 736–742.
12. BertinJ, NirWJ, FischerCM, TayberOV, ErradaPR, et al. (1999) Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem 274 : 12955–12958.
13. ParkJH, KimYG, McDonaldC, KannegantiTD, HasegawaM, et al. (2007) RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol 178 : 2380–2386.
14. Munoz-PlanilloR, KuffaP, Martinez-ColonG, SmithBL, RajendiranTM, et al. (2013) K(+) Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 38 : 1142–1153.
15. MeixenbergerK, PacheF, EitelJ, SchmeckB, HippenstielS, et al. (2010) Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol 184 : 922–930.
16. WitzenrathM, PacheF, LorenzD, KoppeU, GutbierB, et al. (2011) The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 187 : 434–440.
17. CassonCN, CopenhaverAM, ZwackEE, NguyenHT, StrowigT, et al. (2013) Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9: e1003400.
18. BauernfeindFG, HorvathG, StutzA, AlnemriES, MacDonaldK, et al. (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183 : 787–791.
19. LamkanfiM, KannegantiTD (2010) Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol 42 : 792–795.
20. TschoppJ, SchroderK (2010) NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10 : 210–215.
21. KannegantiTD, LamkanfiM, NunezG (2007) Intracellular NOD-like receptors in host defense and disease. Immunity 27 : 549–559.
22. KannegantiTD, OzorenN, Body-MalapelM, AmerA, ParkJH, et al. (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440 : 233–236.
23. ManjiGA, WangL, GeddesBJ, BrownM, MerriamS, et al. (2002) PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem 277 : 11570–11575.
24. FinkSL, CooksonBT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73 : 1907–1916.
25. MiaoEA, LeafIA, TreutingPM, MaoDP, DorsM, et al. (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11 : 1136–1142.
26. KayagakiN, WarmingS, LamkanfiM, Vande WalleL, LouieS, et al. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479 : 117–121.
27. WangS, MiuraM, JungYK, ZhuH, LiE, et al. (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92 : 501–509.
28. CaseCL, KohlerLJ, LimaJB, StrowigT, de ZoeteMR, et al. (2013) Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A 110 : 1851–1856.
29. BrozP, RubyT, BelhocineK, BouleyDM, KayagakiN, et al. (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490 : 288–291.
30. GurungP, MalireddiRK, AnandPK, DemonD, WalleLV, et al. (2012) Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein - and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287 : 34474–34483.
31. RathinamVA, VanajaSK, WaggonerL, SokolovskaA, BeckerC, et al. (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150 : 606–619.
32. MariathasanS, NewtonK, MonackDM, VucicD, FrenchDM, et al. (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430 : 213–218.
33. MartinonF, BurnsK, TschoppJ (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10 : 417–426.
34. FranchiL, AmerA, Body-MalapelM, KannegantiTD, OzorenN, et al. (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol 7 : 576–582.
35. MiaoEA, Alpuche-ArandaCM, DorsM, ClarkAE, BaderMW, et al. (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7 : 569–575.
36. LamkanfiM, MuellerJL, VitariAC, MisaghiS, FedorovaA, et al. (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187 : 61–70.
37. NakahiraK, HaspelJA, RathinamVA, LeeSJ, DolinayT, et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12 : 222–230.
38. ZhouR, YazdiAS, MenuP, TschoppJ (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469 : 221–225.
39. LupferC, ThomasPG, AnandPK, VogelP, MilastaS, et al. (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14 : 480–488.
40. KabeyaY, MizushimaN, UenoT, YamamotoA, KirisakoT, et al. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19 : 5720–5728.
41. LoYY, WongJM, CruzTF (1996) Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem 271 : 15703–15707.
42. YangS, WangB, HumphriesF, JacksonR, HealyME, et al. (2013) Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol 14 : 927–936.
43. MeunierE, DickMS, DreierRF, SchurmannN, Kenzelmann BrozD, et al. (2014) Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509 : 366–370.
44. AachouiY, LeafIA, HagarJA, FontanaMF, CamposCG, et al. (2013) Caspase-11 protects against bacteria that escape the vacuole. Science 339 : 975–978.
45. PillaDM, HagarJA, HaldarAK, MasonAK, DegrandiD, et al. (2014) Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A 111 : 6046–6051.
46. KayagakiN, WongMT, StoweIB, RamaniSR, GonzalezLC, et al. (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341 : 1246–1249.
47. HagarJA, PowellDA, AachouiY, ErnstRK, MiaoEA (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341 : 1250–1253.
48. LiuZ, ZakiMH, VogelP, GurungP, FinlayBB, et al. (2012) Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem 287 : 16955–16964.
49. AkhterA, CautionK, Abu KhweekA, TaziM, AbdulrahmanBA, et al. (2012) Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37 : 35–47.
50. MonackDM, HershD, GhoriN, BouleyD, ZychlinskyA, et al. (2000) Salmonella exploits caspase-1 to colonize Peyer's patches in a murine typhoid model. J Exp Med 192 : 249–258.
51. KovarovaM, HeskerPR, JaniaL, NguyenM, SnouwaertJN, et al. (2012) NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189 : 2006–2016.
52. WillinghamSB, AllenIC, BergstralhDT, BrickeyWJ, HuangMT, et al. (2009) NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 183 : 2008–2015.
53. AnandPK, TaitSW, LamkanfiM, AmerAO, NunezG, et al. (2011) TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J Biol Chem 286 : 42981–42991.
54. BertrandMJ, DoironK, LabbeK, KornelukRG, BarkerPA, et al. (2009) Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30 : 789–801.
55. MariathasanS, WeissDS, NewtonK, McBrideJ, O'RourkeK, et al. (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440 : 228–232.
56. Said-SadierN, PadillaE, LangsleyG, OjciusDM (2010) Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 5: e10008.
57. HugotJP, ChamaillardM, ZoualiH, LesageS, CezardJP, et al. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411 : 599–603.
58. ReingoldL, RahalK, Schmiedlin-RenP, RittershausAC, BenderD, et al. (2013) Development of a peptidoglycan-polysaccharide murine model of Crohn's disease: effect of genetic background. Inflamm Bowel Dis 19 : 1238–1244.
59. JamonttJ, PetitS, ClarkN, ParkinsonSJ, SmithP (2013) Nucleotide-binding oligomerization domain 2 signaling promotes hyperresponsive macrophages and colitis in IL-10-deficient mice. J Immunol 190 : 2948–2958.
60. Couturier-MaillardA, SecherT, RehmanA, NormandS, De ArcangelisA, et al. (2013) NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123 : 700–711.
61. WatanabeT, KitaniA, MurrayPJ, WakatsukiY, FussIJ, et al. (2006) Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity 25 : 473–485.
62. AmendolaA, ButeraA, SanchezM, StroberW, BoirivantM (2013) Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol 7 : 391–404.
63. KobayashiK, InoharaN, HernandezLD, GalanJE, NunezG, et al. (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416 : 194–199.
64. KobayashiKS, ChamaillardM, OguraY, HenegariuO, InoharaN, et al. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307 : 731–734.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- The Secreted Peptide PIP1 Amplifies Immunity through Receptor-Like Kinase 7
- Symbionts Commonly Provide Broad Spectrum Resistance to Viruses in Insects: A Comparative Analysis of Strains
- MIF Contributes to Associated Immunopathogenicity Development
- The Ins and Outs of Rust Haustoria