
The Transcriptional Activator LdtR from ‘ Liberibacter asiaticus’ Mediates Osmotic Stress Tolerance
The rapid expansion of Huanglongbing disease (HLB) has caused a severe crisis in the citrus industry, with no solution visible in the near future. The causative agent, ‘Candidatus Liberibacter asiaticus’, is an unculturable bacterium under common laboratory conditions, which has made it difficult to gain understanding of this pathogen. Here we used a biochemical approach to identify new chemicals that could be used for the treatment of this devastating disease. These chemicals target a specific transcription factor (LdtR) in ‘Ca. Liberibacter asiaticus’. When bound to LdtR, the chemicals inactivate the protein, which disrupts a cell wall remodeling process that is critical for survival of the pathogen when exposed to osmotic stress (i.e. within the phloem of a citrus tree). Several model strains were used to confirm that the newly identified transcription factor (LdtR) and its regulated genes (ldtR and ldtP) confer tolerance to osmotic stress. The results presented in this study provide strong proof of concept for the use of small molecules that target LdtR, as a potential treatment option for Huanglongbing disease.
Published in the journal:
The Transcriptional Activator LdtR from ‘ Liberibacter asiaticus’ Mediates Osmotic Stress Tolerance. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004101
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004101
Summary
The rapid expansion of Huanglongbing disease (HLB) has caused a severe crisis in the citrus industry, with no solution visible in the near future. The causative agent, ‘Candidatus Liberibacter asiaticus’, is an unculturable bacterium under common laboratory conditions, which has made it difficult to gain understanding of this pathogen. Here we used a biochemical approach to identify new chemicals that could be used for the treatment of this devastating disease. These chemicals target a specific transcription factor (LdtR) in ‘Ca. Liberibacter asiaticus’. When bound to LdtR, the chemicals inactivate the protein, which disrupts a cell wall remodeling process that is critical for survival of the pathogen when exposed to osmotic stress (i.e. within the phloem of a citrus tree). Several model strains were used to confirm that the newly identified transcription factor (LdtR) and its regulated genes (ldtR and ldtP) confer tolerance to osmotic stress. The results presented in this study provide strong proof of concept for the use of small molecules that target LdtR, as a potential treatment option for Huanglongbing disease.
Introduction
The rapid expansion of Huanglongbing (HLB; also known as “citrus greening”) disease caused a crisis in the citrus industry worldwide, with no solution visible in the near future. Experts estimate that without pro-active measures, the citrus industry in affected areas (like Florida) will be significantly reduced within 2–10 years. As such, it is critical to further our understanding of the metabolic and regulatory pathways in the causal agent ‘Candidatus Liberibacter asiaticus’ (‘Ca. L. asiaticus’), to facilitate the discovery of new means of prevention and/or treatment for HLB. Various treatment methods, including large scale field applications of penicillin and streptomycin, have been thoroughly examined and resulted in little success [1]. Although not applicable to field studies, thermotherapy (incubation of living plants in chambers at 40°C for 48 h) has been proposed for use in nurseries [2]. Despite all these efforts, current methods to control the spread of HLB are still limited to the removal and destruction of infected trees.
The causal agent of this devastating disease, ‘Ca. L. asiaticus’, is an unculturable bacterium. The inability to culture these species has greatly hindered progress toward the identification of therapeutic targets, and the development of viable treatment options. Furthermore, comparative genome analyses did not identify genes with predicted virulence functions (toxins), or specialized secretion systems (pathogenicity determinants) in the genome of ‘Ca. L. asiaticus’. These analyses did, however, provide valuable insight into the putative mechanisms of gene regulation.
Transcription factors, as defined by the Cluster of Orthologous Groups, constitute less than 2% of the ‘Ca. L. asiaticus’ genome, while in S. meliloti, another member of the Rhizobiaceae family, it comprises 6% of the genome. As a consequence, a small number of transcription factors may control several metabolic pathways. Therefore, we hypothesized that inactivation of a single transcription factor could result in pleiotropic effects, including decreased persistence within the host. CLIBASIA_01180 (renamed LdtR), is a homolog of the multidrug resistance regulator MarR. This regulator is encoded upstream of CLIBASIA_01175, a predicted L,D transpeptidase (renamed LdtP) involved in cell wall remodeling.
Peptidoglycan (PG) modifications have been observed in Gram-positive and Gram-negative bacteria, and often occur in response to environmental changes. The bacterial pathogens Neisseria gonorrhoeae and Listeria monocytogenes modify their PG residues to evade detection by the host immune system, and increase tolerance to stress [3], [4]. The PG structure consists of alternating N-acetylglucosamine (NAG) and β-(1-4)-N-acetylmuramic acid (NAM) residues. A peptide stem linked to the NAM residue mediates the cross-link to other units in the growing PG, forming a three-dimensional mesh-like architecture that confers structural strength and rigidity to the cell wall [5]. The PG of A. tumefaciens and S. meliloti is highly cross-linked (64%), with the muropeptide NAM-L-alanine, D-glutamic acid, DL-diaminopimelic acid, D-alanine being the most frequent [6].
The goal of this study was to characterize and assess the biological importance of LdtR and LdtP, and their role in the persistence of ‘Ca. L. asiaticus’ within citrus hosts. We used a biochemical approach to identify small molecules that modulate the expression and activity of the LdtR transcription factor. As ‘Ca. L. asiaticus’ is yet to be cultured, we used two of its closest culturable phylogenetic relatives, Sinorhizobium meliloti and Liberibacter crescens, as models to assess the biological role of LdtR and LdtP. We also developed a model using ‘Ca. L. asiaticus’ infected shoots, to validate LdtR as an effective target for the design of new therapeutics.
Results
LdtR binds to its own promoter region and to the ldtP promoter region
The ldtR gene encodes the only MarR family member of transcriptional regulators in the genome of ‘Ca. L. asiaticus’ psy62 (ldtRLas). It shares high amino acid sequence identity to proteins found in all Rhizobiaceae family, including: ‘Ca. L. solanacearum’ CLso-ZC1 (89%), Liberibacter crescens BT-1 (73%), Sinorhizobium meliloti 1021 (70%), Agrobacterium tumefaciens F2 (74%), A. radiobacter K84 (71%), Rhizobium leguminosarum bv. viciae 3841 (71%), and Hoeflea phototrophica DFL-43 (65%). The genomic arrangement of ldtRLas was also similar to that of its orthologs (Fig. 1); however, none of these proteins has previously been characterized.

ldtRLas is encoded by the minus strand. CLIBASIA_01185 is encoded 341 bp upstream of ldtRLas, on the plus strand. This gene encodes for a putative delta-aminolevulinic acid dehydratase (hemB) involved in tetrapyrrole biosynthesis [7]. Downstream of ldtRLas, on the minus strand, is ldtPLas, which contains both a YkuD L,D-transpeptidase domain (pfam03734) and a peptidoglycan binding domain (pfam01471), suggesting that it likely acts as an L,D-transpeptidase. Biotinylated probes were generated to contain the intergenic region of CLIBASIA_01185 and ldtRLas (PldtR: −395 to +47, positions are relative to ldtRLas translation start site), as well as the putative promoter region of ldtPLas (PldtP: −248 to +79, relative to the ldtPLas translation start site). EMSA analysis of the interaction between LdtRLas and PldtR or PldtP, revealed higher binding affinity for PldtP, with 50% binding achieved at 100 nM (Fig. 2A). With increasing concentrations of LdtRLas, a higher molecular weight oligomer was also observed. Size exclusion chromatography indicated that LdtRLas is a stable dimer in solution with an observed molecular weight of 39 kDa (Fig. S1). Taken together, these results suggest that there is either a second binding site within the ldtP promoter, or LdtRLas may further oligomerize upon binding to DNA.

To confirm the location of LdtR binding, competitor experiments were conducted using unlabeled DNA probes (Table 1). The largest probe (CD-1) contains the whole sequence used in EMSA (from −248 to +79). Probe CD-2 contains LdtRLas binding site surrounded by promoter elements (−139 to +79). Probe CD-3 was designed to contain only the protected site I identified by DNase I footprinting (−118 to −74), while probe CD-4 does not contain the LdtRLas binding site (−21 to +58). The addition of probe CD-1 or CD-2 resulted in a similar decrease in the intensity of the shifted bands (Fig. 2B). This effect was further enhanced in the presence of probe CD-3. No competition was observed with probe CD-4. These results indicate that LdtRLas may have two binding sites within the ldtP the promoter.

The DNA binding sequence for LdtRLas in the promoter region of ldtPLas was identified by DNase I footprinting. The protected site consists of 18 nucleotides (ATATTCCTTGTATTTTAA, ldtP_1) on the minus strand (Fig. 2C), upstream of the predicted −35 box. Immediately downstream from the protected site, a 15 nt DNase I-hypersensitivity region was identified, which may correspond to a DNA bending site (Fig. 2C). Analysis of the DNA sequence upstream of the hypersensitivity region indicated the presence of a second binding site; however, the binding sequence is broken into two segments separated by 9 nt (ATATTTCTT-n9-GTGATTTAA, ldtP_2; Fig. 2D). A putative binding site was identified in the promoter region of ldtRLas with a similar disruption (ldtR_1, Fig. S2E). This sequence displays a separation of 6 nt between each segment, which may explain the lower affinity of LdtRLas for PldtR (Fig. 2A).
To determine the residues required for LdtR binding, the three binding sites (ldtP_1, ldtP_2, and ldtR_1) were compared, and a position specific frequency matrix was constructed (Fig. 3A). Using plasmid pBS6 as a template, site directed mutagenesis was performed on residues that showed high conservation as follow: M1 (−111)AT→GG, M2 (−108)TT→GG, M3 (−104)TT→GG, M4 (−102)GT→AG, and M5 (−99)TtT→GtG. EMSA experiments were then conducted with the mutated binding sites. Decreased LdtRLas binding was observed with probes PldtP_M1 and PldtP_M5 (Fig. 3B). Mutations M2, M3, and M4 did not affect LdtRLas binding (Fig. 3B).

LdtRLas is a transcriptional activator
To determine the mode of regulation for LdtRLas, we generated several lacZ fusions using Bacillus subtilis as a model strain, since all the genes under study are absent from its genome. This system allows the study of transcriptional fusions by inserting a single copy of the gene into a non-essential chromosomal locus (thrC). The putative promoter regions of CLIBASIA_01185, ldtRLas, and ldtPLas were fused to the lacZ gene, resulting in strains BS1 (PCLIBASIA_01185), BS3 (PldtR) and BS5 (PldtP). All three promoters were found to have very low activity (1.1±0.7, 0.1±0.03 and 3.8±0.07 AU, respectively; Fig. 4). In the presence of ldtRLas, increased expression of lacZ was observed in strains BS4 (PldtR-ldtRLas) and BS6 (PldtR-ldtRLas-PldtP) (16.4±0.02 and 36.3±0.2 AU, respectively; Fig. 4). No expression was observed in strain BS2 (harboring ldtRLas and PCLIBASIA_01185). These results confirmed that LdtRLas is a transcriptional activator of ldtRLas and ldtPLas, while it does not regulate CLIBASIA_01185.

The in vivo specificity of LdtRLas binding to PldtR-ldtRLas-PldtP was tested in strains BS6M1 and BS6M5, harboring mutations M1 or M5 on the PldtP binding site 1. β-galactosidase activity was significantly reduced (p<0.001) by 55% and 47%, for BS6M1 and BS6M5 respectively, when compared to the wild type promoter (Fig. 4). These results positively correlated with the reduced binding of LdtRLas to probes PldtP_M1 and PldtP_M5 in EMSA experiments (Fig. 3B), confirming the specificity of the LdtRLas binding site.
ldtR-ldtP mutations resulted in shortened cells and increased sensitivity to osmotic stress in S. meliloti
Inactivation of L,D-transpeptidases have been shown to induce morphological changes, resulting in decreased rigidity of the cell wall [8]. As ‘Ca. L. asiaticus’ has yet to be cultured, a model strain was used to study the biological role of LdtR and LdtP. Due to its close phylogenetic relationship to ‘Ca. L. asiaticus’, and the availability of genetic tools, S. meliloti was chosen. Prior to in vivo experiments, SMc01768 (named LdtRSmc) was purified and confirmed to bind to its own promoter region, as well as to the promoter region of the ldtPLas homolog, SMc01769 (named LdtPSmc; Fig. S3).
Insertional mutants of ldtPSmc and ldtRSmc were constructed in S. meliloti (strains SMP1 and SMP2, respectively; Table 2) by homologous insertion of pSMP1 and pSMP2 in ldtPSmc and ldtRSmc, respectively. In strain SMP1, ldtPSmc was disrupted at 498 nt from the ATG start codon. In strain SMP2, ldtRSmc was disrupted 29 nt from ATG start codon.

Analysis of crystal violet-stained cells revealed the SMP1 and SMP2 mutants had a shortened rod-type phenotype (short-cell), when compared to the wild type S. meliloti. However, they did not show growth defects in liquid cultures (doubling time or final OD600, data not shown). Scanning electron microscopy was used to verify and quantify these morphological changes. Electron micrographs confirmed the average length of SMP1 (1.16 µm±0.15) and SMP2 (1.15 µm±0.14) mutants to be significantly shorter (30%, p<0.005) than wild type cells (1.65 µm±0.20) (Fig. 5A–C).

To determine if modifications in the cell wall composition would affect tolerance to osmotic stress, a seven-fold serial dilution of each strain was spot plated in the presence of sucrose (0.3 M) or NaCl (0.4 M). Increased sensitivity to osmotic stress was observed in strain SMP1 (1.8×107±3.9×106 and 1.4×106±5.4×105 CFU/ml, for sucrose and NaCl respectively), and SMP2 (1.6×107±7.1×105 and 1.1×106±7×105 CFU/ml, for sucrose and NaCl respectively), when compared to the wild type strain (2.2×108±2.5×107 and 7.1×106±6.9×105 CFU/ml, for sucrose and NaCl respectively; Fig. 6A). These results were significantly different for sucrose (p<0.05) but not for NaCl. Higher concentrations of NaCl or sucrose were toxic for all strains (data not shown).

To establish a link between elevated sensitivity to osmotic stress, and the regulation of gene expression by LdtRSmc, β-glucuronidase activity (encoded by the uidA gene) was measured in S. meliloti (Fig. 6B). Strain SMP3 was constructed by inserting the uidA reporter gene downstream of ldtPSmc (no disruption to ldtRSmc or ldtPSmc). Strain SMP3 was used as a reporter strain to determine the expression of uidA in a wild type phenotype. In the presence of NaCl, the β-glucuronidase activity was induced in a concentration-dependent manner in strains SMP3 and SMP1 (Fig. 6B). Induction of β-glucuronidase activity was dependent on the presence of LdtRSmc. In absence of the regulator (strain SMP2), no changes in the expression of the reporter gene were observed. These results confirm the role of LdtRSmc as an activator of ldtRSmc and ldtPSmc transcription in response to osmotic stress.
To determine if the tolerance to osmotic stress could be recovered by the addition of ldtR, strain SMP2 (ldtR mutant) was transformed with plasmid pSMP4 carrying ldtRLas (strain SMP2B), and analyzed for sensitivity to osmotic stress. Strain SMP2A (carrying the empty pBBR1MCS-5 plasmid [9]) served as a control. Increased tolerance to osmotic stress was observed in strain SMP2B (6.1×106±5.7×105 and 3.6×106±7.6×105 CFU/ml, for sucrose and NaCl respectively p<0.05), when compared to SMP2A (7.2×105±5.8×104 and 5.7×105±1.0×105 CFU/ml, for sucrose and NaCl respectively) (Fig. 6C). These results suggest that LdtRLas is directly involved in tolerance to osmotic stress by recognizing similar promoter elements in PldtP of S. meliloti. Further in silico analyses in S. meliloti revealed the presence of LdtRLas binding sites upstream of the ldtP −35 sequence, in agreement with the arrangement of LdtR binding sites in L. crescens and ‘Ca. L. asiaticus’ (Fig. S2).
To determine if the addition of ldtPLas could recover the tolerance to osmotic stress, strain SMP2 (ldtR mutant) was transformed with pSMP5 carrying ldtPLas (SMP2C). Increased tolerance to osmotic stress was observed in strain SMP2C (1.5×107±2.6×106 and 8.0×106±2.1×106 CFU/ml, for sucrose and NaCl respectively, p<0.05) when compared to SMP2A (7.2×105±5.8×104 and 5.7×105±1.0×105 CFU/ml, for sucrose and NaCl respectively, (Fig. 6C). These results indicate that LdtP from S. meliloti and ‘Ca. L. asiaticus’ are functionally homologous. Taken together these findings confirm that the decreased tolerance to osmotic stress observed in strain SMP2, was due to the absence of LdtRSmc transcriptional activity.
Identification of small molecules that modulate the activity of LdtRLas
A fluorescence based small molecule screening assay [10] was used to identify chemical scaffolds that may interact with the transcription factor LdtRLas. We utilized a library containing 196 biologically relevant small molecules [11], [12] and the Prestwick Chemical Library, which contains 1,200 small molecules [10]. Small molecules that induced a shift in the melting temperature (ΔTm) of LdtRLas, by more than two degrees, were considered as positive “hits”. The chemicals with the strongest destabilizing effect were hexestrol (ΔTm = −2.5±0.5°C), diethylstilbestrol (ΔTm = −4.5±0.9°C), and benzbromarone (ΔTm = −2.0±0.3°C), while oxantel pamoate (ΔTm = 2.0±0.2°C) was found to greatly increase the stability of the protein (Fig. S4).
Small molecules decrease LdtRLas binding to PldtP
A change in thermal stability does not guarantee a biologically relevant interaction; therefore, each of the compounds was tested on the ability to modulate the PldtP: LdtRLas interaction. All of the identified chemicals decreased the DNA binding activity of LdtRLas in a concentration-dependent manner (Fig. 7). Benzbromarone had the strongest effect and disrupted the PldtP: LdtRLas interaction at 50 µM. Oxantel pamoate completely impaired the PldtP: LdtRLas interaction at 250 µM, where only partial disruption of the complex was observed with hexestrol and diethylstilbestrol. The chemical scaffold of the strongest destabilizing agents (benzbromarone and hexestrol) served to identify other natural compounds such as resveratrol and phloretin. It was found that resveratrol decreased binding at 250 µM, while phloretin disrupted the PldtP: LdtRLas interaction at 100 µM, consistent with molecules having physiological relevance (Fig. 7). To determine the specificity of each ligand that decreased PldtP: LdtRLas interaction, EMSA experiments were carried using a MarR homolog (LVIS0553), in the presence or absence of each chemical. The PLVIS0553: LVIS0553 interaction was previously found to be modulated by the presence of novobiocin [10]. As expected none of the identified ligands for LdtRLas affected the binding of LVIS0553 to its cognate promoter (Fig. S5).

Small molecules induce morphological changes in S. meliloti and L. crescens
We hypothesized that chemicals that modulate binding of the transcription factor would result in phenotypic abnormalities, similar to those observed in ldtR mutants of S. meliloti. The toxicity of each chemical was determined and sub-lethal concentrations were used for these experiments (Table 3). As expected, the addition of increasing concentrations of each chemical (25 µM phloretin, 25 µM benzbromarone, or 1 µM hexestrol) resulted in a pronounced decrease in cell size in S. meliloti (Fig. 8A). Quantitative assessments of the cell size were conducted in wild type S. meliloti cells grown in the presence of 25 µM phloretin. The addition of phloretin resulted in a significant decrease of 27% in the cell size (1.20 µm±0.18, p<0.005; Fig. 5D) when compared to the wild type (1.65 µm±0.20; Fig. 5A). These results are in agreement with the decrease in cell size observed for the SMP1 and SMP2 mutants (Fig. 5B and C).

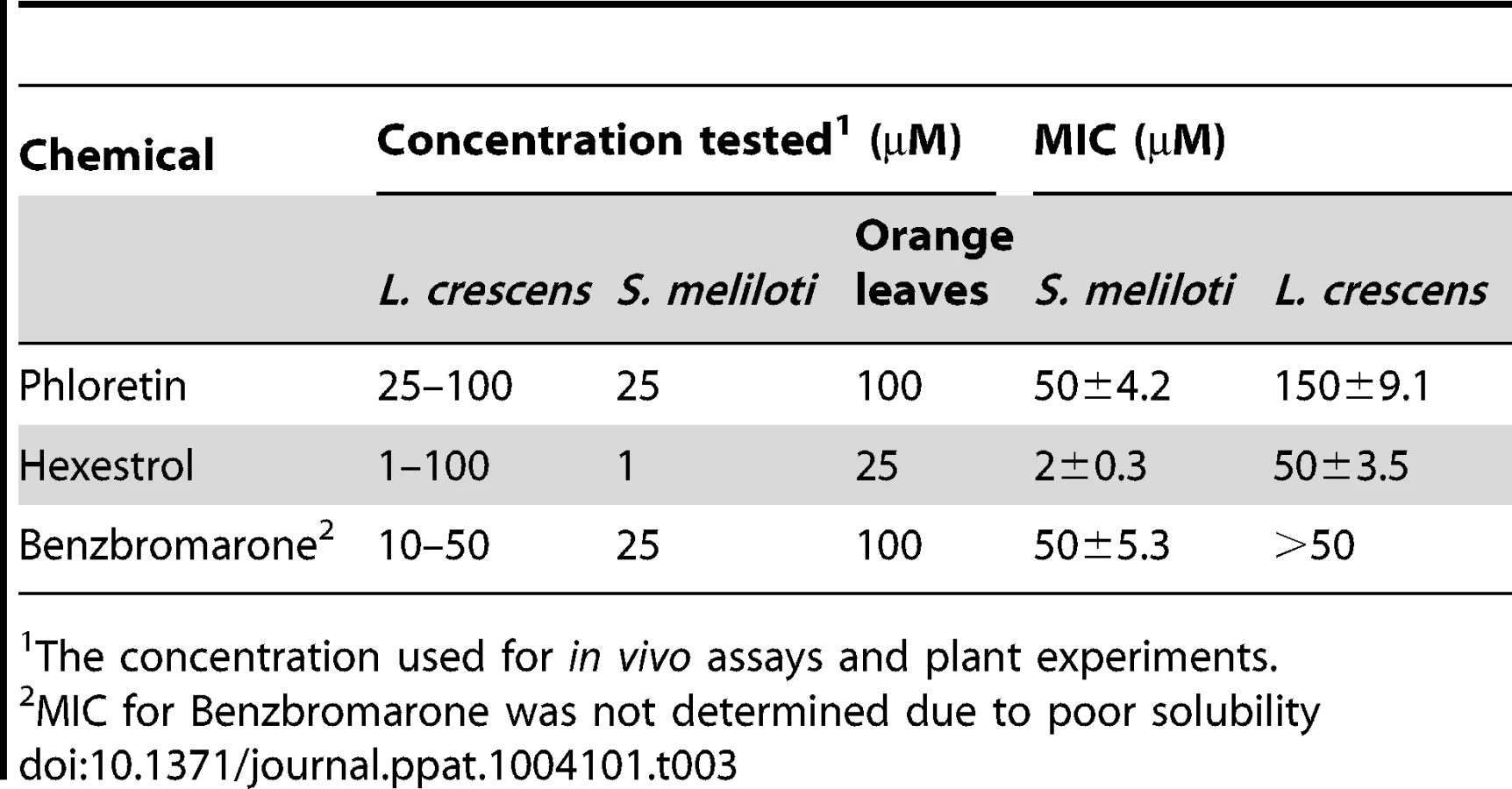
Confirmatory studies were performed in L. crescens BT-1. L. crescens is a close relative of ‘Ca. L. asiaticus’ that was recently isolated from mountain papaya, and can be cultured under laboratory conditions. In addition, the complete genome of L. crescens BT-1 has been sequenced [13], and the homolog of ldtRLas (B488_10910, named ldtRLcr) identified. The chemicals (50 µM phloretin, 50 µM benzbromarone, or 25 µM hexestrol) that induced the “short-cell” phenotype in S. meliloti modulated the activity of ldtRLcr, resulting in a similar phenotype in L. crescens BT-1 (Fig. 8B).
To test if the phenotype induced by the presence of the chemicals correlated with changes in the expression of the ldtRLcr and B488_10900 (named ldtPLcr), the mRNA levels were determined. L. crescens was grown to exponential phase, in presence or absence of the small molecules. Modest, but highly reproducible decreases of 45.4±8.9, 62.5±7.7, and 37.5±11.5 percent in ldtPLcr expression, were observed upon growth in the presence of 25 µM phloretin, 50 µM benzbromarone, or 25 µM hexestrol, respectively. These results confirmed the role of the small molecules in modulating the activity of LdtRLcr, in vivo.
Small molecules decreased stress tolerance in S. meliloti and L. crescens
Based on the pivotal role of peptidoglycan in counteracting the effects of osmotic pressure, we hypothesized that the downregulation of ldtR and ldtP, by chemicals that impair LdtR activity, will result in decreased tolerance to osmotic stress.
The S. meliloti wild type-phenotype strain, SMP3, was used to evaluate the effect of the small molecules, on the ability to grow under osmotic stress conditions. The cells were grown in the presence or absence of phloretin or benzbromarone with increasing concentrations of NaCl (Table 3; Fig. 9A). In the presence of the small molecules, strain SMP3 showed a severe decrease in tolerance to NaCl. At NaCl concentrations as low as 50 mM, a decrease in growth was observed in the presence of phloretin or benzbromarone (50 and 30%, respectively). Under these conditions, β-glucuronidase activity was determined. Induction of ldtRSmc and ldtPSmc expression, in response to high concentrations of NaCl, was overturned in presence of phloretin or benzbromarone (Fig. 9B). These results are in agreement with the decrease tolerance to osmotic stress observed in presence of the small molecules.

Since genetic tools are not available yet to manipulate L. crescens, we determined the effect of the addition of chemicals, at sublethal concentrations, on the ability to tolerate high concentrations of NaCl or sucrose. It was establish that the maximal concentration of NaCl and sucrose that L. crescens tolerate is 150 and 200 mM, respectively (Fig. S6). The effect of increasing concentrations of the small molecules was tested on the ability to tolerate NaCl or sucrose. The addition of phloretin, benzbromarone, or hexestrol (50, 100, or 25 µM, respectively), did not affect the growth of L. crescens in control conditions. Conversely, in the presence of NaCl or sucrose, L. crescens displayed increased sensitivity to all chemicals tested (Fig. 10). Together, these results indicate that in S. meliloti and L. crescens, tolerance to osmotic stress is in part mediated by changes in the peptidoglycan crosslinking, which can be manipulated by the addition of small molecules that modulate mRNA levels through LdtR activity.

Small molecules as therapeutics
Based on these results, we designed an in vitro model to test the effectiveness of these chemicals. Shoots were collected from a single HLB-symptomatic Valencia Orange (C. sinensis) tree, infected with ‘Ca. L. asiaticus’. Previous studies have reported greater numbers of viable ‘Ca. L. asiaticus’ cells in the sieve elements of young, asymptomatic leaves, collected from new flushes [14]. All leaves used for this study were collected from new flushes on highly symptomatic branches. Nine leaves were collected for each treatment and control group. Samples were then incubated for 6 or 24 h (with or without chemical).
Since ‘Ca. L. asiaticus’ still remains elusive to culture under laboratory conditions, we followed the transcriptional activity of the 16S RNA gene and the L10 ribosomal protein (encoded by the rplJ gene) as viability parameters. The amplification values were normalized to the plant gene cox2 and are expressed relative to the control (incubated without chemical) samples. After 24 h of incubation, significant differences were observed in samples treated with small molecules. Expression of the 16S RNA gene was repressed in samples treated with hexestrol and phloretin [39.7±9.8 (p<0.05) and 55.9±9.5 (p<0.005) percent decrease, respectively], while benzbromarone showed the strongest effect, with 90.9±6.1 percent decreased expression (p<0.005) (Fig. 11A). A similar trend was observed for the expression of rplJ, with a decreased expression of 94.2±2.3, 94.6±2.9, and 97.6±1.5 percent for phloretin, hexestrol, and benzbromarone, respectively (p<0.005) (Fig. 11B). After a short period of incubation (6 h) no significant changes were observed (data not shown).

The effect of the chemicals on the expression of the specific genes ldtRLas and ldtPLas was then determined in the infected leaves. The expression values are calculated relative to the 16S RNA gene, to assess the specificity of the chemicals to target genes. Phloretin showed a strong effect (88.1±1.2 percent decrease) on the expression of ldtRLas after 6 h of incubation, while benzbromarone displayed similar decreased expression values after 6 or 24 h (78.9±1.3 and 80.5±1.1 percent, respectively, p<0.005; Fig. 11C). The expression of ldtPLas showed constant and incremental repression values over time. Hexestrol and benzbromarone reached maximal values of 93.7±0.8 and 94.2±0.9, respectively, while phloretin showed a maximal value of 84.8±3.8 percent decrease (p<0.005) (Fig. 11D).
These results indicate that the small molecules tested act specifically on the ldtRLas activator. We hypothesize that in ‘Ca. L asiaticus’, expression of LdtP is increased in response to osmotic stress, allowing persistence of the bacteria within the phloem of the tree. As such, the regulation of ldtP expression through inactivation of LdtR with small molecules represents a direct means of influencing osmotic stress tolerance, and survival of ‘Ca. L asiaticus’ within the host.
Discussion
‘Ca. L. asiaticus’ is frequently exposed to changes in osmotic pressure, due to variations in phloem sap composition. Sucrose concentrations in the phloem can vary significantly (between 0.5 and 30% w/v, corresponding to 15 mM and 880 mM, respectively) depending on plant species, tissue, time of day, and season [15], [16]. Consequently, bacterial pathogens that replicate in the phloem must continuously respond to changes in osmotic pressure. In this context, L,D transpeptidase activity is critical, as these enzymes are directly involved in cell wall biosynthesis and remodeling in response to stress conditions.
In this report, we identified and characterized a regulon from the citrus pathogen ‘Ca. L. asiaticus’, involved in peptidoglycan remodeling. These results represent the first regulatory system functionally analyzed for this pathogen. Included in this regulon is ldtR, a member of the MarR family of transcriptional regulators, and ldtP, a predicted L,D-transpeptidase. The genomic context of ldtRLas was conserved among members of the Rhizobiacea family. As such, the two closest phylogenic relatives of ‘Ca. L. asiaticus’, S. meliloti and L. crescens, were used to study the phenotypic effects of L,D-transpeptidase inactivation, and the physiological conditions that contribute to the expression of the ldtR regulon, since ‘Ca. L. asiaticus’ is yet to be cultured. The highly conserved nature of ldtR suggests a similar mechanism of regulation among these members of the Rhizobiacea family; however, the response to ligands may vary due to the different lifestyle of each species.
L,D-transpeptidases (E.C. 2.3.2.12) mediate the substitution of 4→3 (D-Ala4 to mDAP3) crosslinks, generated by the penicillin binding protein D,D-transpeptidase, to 3→3 (mDAP3 to mDAP3) crosslinks. This pattern of L,D-transpeptidation represents 80% of the crosslinks observed in the cell walls of stationary phase M. tuberculosis cells [17]. Similar results were observed in other microorganisms, including E. coli and V. cholerae [8], [18]. These observations suggest that transpeptidation is an active process in stationary phase cells, which may be critical for adaptation and tolerance to environmental stress. In M. tuberculosis, increased cell wall transpeptidation was positively correlated with increased transcription of LdtM1 during nutrient starvation [17], [19]. Interestingly, our results in L. crescens indicate that ldtPLcr and ldtRLcr are expressed throughout the growth phases, when cultured under laboratory conditions. However, a comparative analysis of the ‘Ca. L. asiaticus’ transcriptome revealed that ldtR expression was five times higher in samples obtained from infected trees, when compared to samples collected from infected psyllids (an alternate host and insect vector of ‘Ca. L. asiaticus’) [20]. These results suggest that in ‘Ca. L. asiaticus’, transcription of Ldt-associated genes may be triggered by the high osmotic pressure generated by the phloem sap. These data, in combination with previous reports of the large proportion of 3→3 crosslinks in the muropeptides of Rhizobiales, suggest that LdtP may be involved in both housekeeping activities and stress response.
To further explore the LdtR regulatory mechanism, Bacillus subtilis was used as a heterologous host. Interestingly, we found that LdtR acts as a transcriptional activator of the ldtR and ldtP genes. Although the majority of MarR proteins act as transcriptional repressors, several MarR transcriptional activators have been described. In S. meliloti, the MarR family member ExpG binds to the ExpADGE operon to activate expression of the galactoglucan biosynthesis genes [21]. Similarly, PntR and PenR, from Streptomyces arenae and S. exfoliatus, respectively, activate synthesis of the pentalenolactone antibiotic [22]. Interestingly, all of these regulators bind AT-rich sequences similar to the binding sequence identified for LdtR [21]–[23]. This high degree of conservation could represent a common feature among binding sequences for MarR members that act as transcriptional activators.
In S. meliloti, changes in cell morphology (short-cell phenotype) were induced by the mutagenesis of ldtR and ldtP. Similar changes in cell morphology have been described for S. meliloti and Rhizobium spp in response to the accumulation of compounds such as glycine, which decreases the extent of crosslinks [24]–[26]. A similar short-cell phenotype was also observed in V. cholerae, following the accumulation of D-amino acids in the media [18]. Analysis of the ‘Ca. L. asiaticus’ genome revealed no homologs of the transpeptidases involved in these activities, however, a glutamate and alanine racemase were identified. These enzymes contribute to fluctuations in the concentration of D-amino acids. The potential involvement of LdtR in the regulation of these genes may explain the phenotypic changes observed in ldtR mutants. The direct or indirect involvement of LdtR in the regulation of these racemases is currently under examination.
Based on the biological relevance of the ldtR regulon, we identified small molecules (phloretin, benzbromarone, and hexestrol) that decreased binding of LdtR to its cognate promoters, resulting in decreased expression of ldtP and ldtR. In L. crescens, decreased gene expression in presence of these small molecules was positively correlated with decreased tolerance to osmotic stress. Furthermore, in S. meliloti, the addition of phloretin, benzbromarone, or hexestrol resulted in morphological changes (short-cell phenotype) similar to those observed in ldtR and ldtP mutants. Consequently, we reasoned that chemical manipulation of LdtRLas activity will reduce long term survival and persistence of the pathogen in infected citrus trees. Thus, we designed an in vitro model using sweet orange leaves infected with ‘Ca. L. asiaticus’, to validate the effect of these chemicals. In samples treated with the small molecules, a significant decrease in ldtR and ldtP expression was observed, confirming the specific effect of these chemicals in ‘Ca. L asiaticus’. The use of a specific target is essential for the development of an effective therapeutic treatment. Modulation of cell wall transpeptidation has been used as a therapeutic treatment for recalcitrant microorganisms, such as Mycobacterium tuberculosis [27]. In contrast, current efforts towards the treatment of Huanglongbing disease are focused primarily on the use of “broad spectrum” treatments (i.e. penicillin, streptomycin, and thermotherapy). This study provides strong proof of concept for the use of small molecules that target LdtRLas, as a potential treatment option for Huanglongbing disease.
Materials and Methods
Strains and growth conditions
Bacterial strains and plasmids are listed in Table 2. Escherichia coli and Bacillus subtilis strains were grown in Luria-Bertani (LB) medium at 37°C. S. meliloti cells were grown at 30°C in either LB medium or M9 minimal medium with glucose. When required, the media was supplemented with gentamicin (30 µg ml−1), ampicillin, (100 µg ml−1), or chloramphenicol (170 µg ml−1) for E. coli; neomycin (100 µg ml−1), gentamicin (30 µg ml−1), and streptomycin (250 µg ml−1) for S. meliloti; or with erythromycin (1 µg ml−1) for B. subtilis.
L. crescens BT-1 was cultured at 25°C, with moderate agitation (150 rpm), in modified BM7 media [13] containing 1% Brain Heart Infusion (Difco Laboratories, Detroit, MI), 15% Fetal Bovine Serum (Sigma-Aldrich, St. Louis, MO), 30% TMN-FH insect medium (Sigma), α-Ketoglutaric acid (2 mg ml−1), ACES (10 mg ml−1), and potassium hydroxide (3.75 mg ml−1), at pH 6.9. Sodium chloride (0–200 mM) or sucrose (0–300 mM) was added to the growth media to induce osmotic stress. All antibiotics and chemicals were purchased from Sigma-Aldrich.
DNA manipulations
Standard methods were used for chromosomal DNA isolation, restriction enzyme digestion, agarose gel electrophoresis, ligation, and transformation [28]. Plasmids were isolated using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA), and PCR products were purified using Qiaquick purification kits (Qiagen).
For protein expression and purification, ldtR gene was amplified from ‘Ca. L. asiaticus’ str. psy62 or S. meliloti 1021 chromosomal DNA via PCR, and then cloned into the p15TV-L plasmid as described previously [10].
Protein purification
Protein expression and purification was performed as previously described [10]. Concisely, the His-tagged fusion proteins were overexpressed in E. coli BL21-Star(DE3) cells (Agilent Technologies, Santa Clara, CA). The cells were grown in LB medium at 37°C to an OD600 = 0.6 and expression induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After addition of IPTG, the cells were incubated with shaking at 15°C overnight. The cells were harvested and resuspended in binding buffer (500 mM NaCl, 5% glycerol, 50 mM Tris pH 8.0, 5 mM imidazole, 0.5 mM TCEP), and stored at −80°C. The thawed cells were lysed and passed through a French Press. The lysate was clarified by centrifugation (30 min at 17,000 rpm at 4°C) and applied to a metal chelate affinity-column charged with Ni2+. After the column was washed, the protein was eluted from the column using elution buffer (binding buffer with 250 mM Imidazole). The hexa-histidine tag was then cleaved from the protein by treatment with recombinant His-tagged TEV protease. The cleaved protein was then resolved from the cleaved His-tag and the His-tagged protease by passing the mixture through a second Ni2+-column. The purified proteins were dialyzed against 10 mM Tris pH 8.0, 500 mM NaCl, 0.5 mM TCEP, and 2.5% glycerol. Finally, the proteins were aliquoted and stored at −80°C.
Small molecule screening by differential scanning fluorimetry
Purified LdtR protein was screened against a library of 160 intracellular compounds [11] at a final concentration of 100 µM, or against the Prestwick chemical library of 1152 compounds (Prestwick Chemical, France) at a final concentration of 1.3 µg/mL, using fluorometry as previously described [11], [29]. LdtR was diluted to a final concentration of 30 µM in 100 mM Tris pH 8.0, 150 mM NaCl. SYPRO orange was added to a final concentration of 5×. 25 µL aliquots of protein solution containing the chemical compounds were placed in duplicate into 96 well plates (Bio-Rad, Hercules, CA) and heated from 25°C to 80°C at the rate of 1°C per minute. A real-time PCR device (iCycler IQ, Bio-Rad) was used to monitor protein unfolding by the increase in the fluorescence of the fluorophor SYPRO Orange (Life Technologies, Grand Island, NY). Fluorescence intensities were plotted against temperature for each sample well and transition curves were fitted using the Boltzmann equation using Origin 8 software (Northampton, MA). The midpoint of each transition was calculated and compared to the midpoint calculated for the reference sample. If the difference between them was greater than 2.0°C, the corresponding compound was considered to be a “hit” and the experiment was repeated to confirm the effect in a dose dependent manner. Figure S4 shows the melting curves obtained for LdtRLas without chemicals or in presence of the selected hit chemicals. The chemicals that were not selected displayed melting curves similar to the one observed for the control.
Electrophoretic Mobility Shift Assays (EMSAs)
Gel shift assays for LdtR were performed using aliquots of protein purified and concentrated according to the procedures described above. Fragments of the ldtR, and ldtP promoters were generated by PCR using biotin prelabeled (5′-end) primers (Table 1), then purified using QIAquick spin columns (Qiagen). Incubation mixtures for EMSA (20 µL) contained 1 ng of a 5′-labelled DNA probe, 50 mM Tris-HCl pH 7.2, 150 mM KCl, 10 mM MgCl2, 0.01% Triton ×100, 12.5 ng/µL of both Poly(dI-dC) and Poly(dA-dT) nonspecific competitor DNAs, purified LdtR protein (0–400 nM), and ligand (0–1 mM) when indicated. After incubation for 20 min at 37°C, samples were separated on 6% acrylamide-bisacrylamide nondenaturing gels in 0.5× Tris borate-EDTA buffer, pH 8.3 (TBE). Electrophoresis was performed at 100 V using ice-cold 0.5× TBE as a running buffer. DNA was then transferred from the polyacrylamide gel to a Hybond-N+ membrane (GE Healthcare, Pittsburgh, PA) by electroblotting at 250 mA for 45 min in a semidry transfer. Transferred DNA was cross-linked for 15 min using a UV cross-linker equipped with 312 nm bulbs. Biotin labeled DNA was detected using a Phototope-Star Detection Kit (New England Biolabs, Ipswich, MA). Membranes were exposed to Kodak X-ray film.
For the EMSA competitions assays, different fragments of the promoter regions were synthesized using PCR, or by annealing of primers as previously described [10] (Table 1).
DNase I footprinting
Protection assays were performed on both minus and plus strands using 5′-6FAM or 5′-VIC labeled probes generated by PCR using primers described in Table 1. The protection assay contained the same components used for EMSAs, except that 5 ng µl−1 PldtP labeled probe, 6 µM LdtRLas, 0.5 mM CaCl2, 2.5 mM MgCl2, and 0.025 U of DNase I (New England Biolabs) were added into 200 µL of reaction. The mix was incubated for 20 min at 37°C, and ended by adding 50 mM EDTA pH 8.0. The corresponding digestion reaction without LdtR was included as a control. The digested DNA and the sequencing reaction products were analyzed at the Plant and Microbe Genomics facility, Ohio State University, Columbus, using a 3730 DNA analyzer. The protected regions were identified using GeneMapper software (Life Technologies), as previously described [30].
5′RACE-PCR
The transcription start site of ldtR and ldtP genes from ‘Ca. L. asiaticus’ and L. crescens were determined by a modified 5′RACE-PCR protocol. Cultures of B. subtilis BS6 (for ‘Ca. L. asiaticus’ ldtR and ldtP) and L. crescens were grown to exponential phase as described above. The total RNA was extracted using the RiboPure-Bacteria kit (Ambion, Austin, TX) following the manufacturer's protocol. 2.5 µg of each RNA was first treated with 20 U of the Calf intestine alkaline phosphatase (New England Biolabs) for 1 h to remove the 5′-PO4 from degraded RNAs followed by a phenol∶chloroform∶isoamylalcohol precipitation. The RNAs were further treated with 2.5 U of Tobacco acid pyrophosphatase (Epicentre Biotechnologies, Madison, WI) for 1 h to remove the 5′-cap from mRNAs. The CIP/TAP RNAs were then ligated to the Oligo_RACE_RNA adapter (Table 1). The synthesis of the first strand of cDNAs were carried out using primers described in Table 1, with the SuperScript II Reverse Transcriptase (Invitrogen) and according to the manufacturer's protocol. The cDNAs were amplified by PCR using Oligo_RACE_Fw and LdtRLas_RACE_Rv or LdtPLas_RACE_Rv for ‘Ca. L. asiaticus’. Similarly, Oligo_RACE_Fw and LdtRLcr_RACE_Rv or LdtPLcr_RACE_Rv were used for L. crescens (Table 1). The PCR fragments were cloned using the StrataClone Blunt PCR cloning kit (Agilent Technologies), following the manufacturer's protocol. The clones were sequenced and ldtR and ldtP transcriptional start sites determined.
Size-exclusion chromatography
100 µl of protein samples were prepared using 10 mM Tris pH 8.0, 500 mM NaCl, and 10 µM LdtRLas. The sample was incubated 20 min on ice and then injected onto a prepacked Superose 12 10/300 GL gel filtration column (GE Healthcare), connected to a LCC-501 plus (GE Healthcare), and equilibrated with 10 mM Tris pH 8.0 and 500 mM NaCl. Filtration was performed in a flow rate of 0.5 ml/min at 4°C. The eluted protein was monitored continuously for absorbance at 280 nm using a UV-M II monitor (GE Healthcare). Blue dextran 2000 was used to determine the void volume of the column. A combination of protein molecular weight standards, including IgG (150 kDa), BSA (66 kDa), Albumin (45 kDa), Trypsinogen (24 kDa), Cytochrome C (12.4 kDa), and Vitamin B12 (1.36 kDa) was also applied to the column under the same conditions. The elution volume and molecular mass of each protein standard was used to elaborate a standard curve for further determination of the molecular weight of the proteins under study. The theoretical molecular weight of LdtR was calculated from the amino acid sequence using the Compute pI/Mw tool at the ExPASy Proteomics Server (http://ca.expasy.org/tools/pi_tool.html).
Construction of lacZ fusions and β-galactosidase assays
Plasmid pDG1663 [31] was used for the transcriptional analysis of ldtR expression. Plasmids pBS1, pBS2, pBS3, pBS4, pBS5, and pBS6 described in Table 2, were constructed using primers listed in Table 1. To this end, the PCR fragments were cut with HindIII and BamHI restriction enzymes, and ligated into pDG1663 previously digested with the same restriction enzymes. The recombinant clones selected in E. coli DH5α were confirmed by sequencing with primer pDGseq9_Fw. Plasmids pBS6M1, pBS6M2, pBS6M3, pBS6M4, and pBS6M5 were constructed by site-directed mutagenesis in pBS6 using the QuikChange Site-directed Mutagenesis kit (Agilent Technologies). The primers used are listed in Table 1. The transfer of plasmids pBS1, pBS2, pBS3, pBS4, pBS5, pBS6, pBS6M1, pBS6M2, pBS6M3, pBS6M4, and pBS6M5 into B. subtilis 168 was carried out by natural competence [32]. The new generated strains are listed and detailed in Table 2. The integration into the thrC locus was confirmed via extraction of B. subtilis genomic DNA using DNeasy Blood and Tissue kit (Qiagen), followed by PCR with primers pDGseq9_Fw and pDGseq10_Rv (Table 1).
For the β-galactosidase assays, B. subtilis cells were grown at 37°C in LB medium until reached an OD600 of 0.3 (mid-exponential phase). Cells were collected and washed twice with 0.9% NaCl, and permeabilized with 1% toluene in Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol) [33]. β-galactosidase activity was assayed by following the catalytic hydrolysis of chlorophenol red-β-D-galactopyranoside (Sigma-Aldrich). The absorbance at 570 nm was read continuously using a Synergy HT 96-well plate reader (BioTek, Winooski, VT). β-galactosidase activity, expressed as arbitrary units (AU), was calculated using the slope of absorbance curve normalized with the initial cell density. The assays were performed in triplicates.
Construction of reporter, gene disruption and complemented strains of S. meliloti and β-glucuronidase assays
Promoter fusions to the uidA reporter gene, as well as ldtRSmc and ldtPSmc disruption mutants, were generated using plasmid pVMG [34]. pVMG is a modified version of plasmid pVO155, containing a multiple cloning site upstream of a promoterless β-glucuronidase (uidA) reporter gene [35]. For the generation of recombinant strains, a ∼400 bp region of the target gene (−378 to +29 of ldtRSmc, +104 to +498 of ldtPSmc, and +932 to +1332 of ldtPSmc, for pSMP2, pSMP1, and pSMP3, respectively) was amplified by PCR using the primers detailed in Table 1. The amplified fragments were inserted into the SpeI and AgeI restriction sites upstream of uidA, in pVMG. The resultant plasmids were propagated in DH5α and mobilized into S. meliloti 1021 via triparental mating, using helper plasmid pRK600 [36]. Transconjugants were selected on M9 sucrose-neomycin plates and their correct insertion confirmed by sequencing, using primers upstream of the original fragment used for cloning into pVMG and primer Gus_Seq_Rv, located 204 bp inside uidA reporter gene (Table 1).
For complementation assays, the complete sequence of ldtRLas gene was amplified by PCR using primers LdtRLas_EcoRI_Fw and LdtRLas_BamHI_Rv, while ldtPLas sequence was amplified using primers LdtPLas_KpnI_Fw and LdtPLas_EcoRI_Rv (Table 1). The DNA fragments were inserted into pBBR1MCS-5 plasmid, previously digested with the corresponding restriction enzymes, generating plasmids pSMP4 (ldtRLas) and pSMP5 (ldtPLas). The recombinant plasmids were selected in DH5α and confirmed by sequencing using universal M13 primers (Table 1). Plasmids pBBR1MCS-5, pSMP4, and pSMP5 were mobilized into S. meliloti SMP2 via triparental mating, using helper plasmid pRK600 [36]. Transconjugants were selected on M9 sucrose-neomycin-gentamicin plates.
For the β-glucuronidase assays, S. meliloti cells were grown in M9 minimal media, supplemented with NaCl, phloretin, or benzbromarone when indicated, until reached late-exponential phase. Cells were collected and washed twice with 0.9% NaCl, and permeabilized with 1% toluene in Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol) as previously described [33]. β-glucuronidase activity was measured by means of the hydrolysis of 4-nitrophenyl β-D-glucuronide substrate (Sigma-Aldrich). The absorbance at 405 nm was read continuously using a Synergy HT 96-well plate reader (BioTek). β-glucuronidase activity was expressed as µM of p-nitrophenol generated per min, normalized with the initial cell density. The assays were performed in triplicates.
qRT-PCR studies
L. crescens cells were cultured in broth with hexestrol (25 µM), phloretin (50 µM), or benzbromarone (50 µM) when required. The cells were collected by centrifugation at 4°C when OD600 = 0.3 (mid-exponential phase). Total RNA was subsequently isolated with RiboPure-Bacteria (Ambion) in accordance with the manufacturer's protocol. cDNAs were synthesized with the Superscript first-strand synthesis kit (Life Technologies) in accordance with the manufacturer's instructions and stored at −80°C prior to use. Real-time quantitative PCR (qRT-PCR) was carried out in a iCycler IQ apparatus (Bio-Rad) using Platinum SYBR Green qPCR SuperMix for iCycler (Life Technologies) in accordance with the manufacturer's recommended protocol. Primers used for the qRT-PCR are described in further detail on Table 1. The RNA polymerase sigma factor rpoD, 50S ribosomal protein L10, 50S ribosomal protein L12 genes, (B488_13350, B488_08460, B488_08450, respectively), and 16S ribosomal RNA were used as internal controls.
Stress resistance assays
To test resistance to NaCl and sucrose, S. meliloti cells were grown in LB media to exponential phase (OD600 = 1.0). Serial dilutions were made and 4 µl was spot plated. Plates were prepared to contain 0.4 M NaCl or 0.3 M sucrose. In L. crescens the effect of chemical inactivation of LdtR on the stress tolerance was tested by following growth (as increased optical density) on liquid cultures.
Scanning Electron Microscopy (SEM)
The morphology of different strains of S. meliloti (Table 2) was visualized by scanning electron microscopy using a Hitachi S-4000 FE-SEM apparatus (ICBR Electron Microscopy Core Lab, University of Florida, FL). S. meliloti 1021 strain, grown in the presence or absence of 25 µM phloretin, as well as SMP1 and SMP2 mutants, were cultured until exponential phase (OD600 = 1.0) in LB media, as described above. Prior fixation, the cells were centrifuged 3 min at 8,000 rpm and the pellets washed twice with 1× PBS buffer. Finally, the cells were treated with 1 mL of Trump's fixative solution for 20 min at room temperature, and post-fixed in 1% osmium tetroxide followed by dehydration in graded ethanol concentrations, following Electron Microscopy Core Lab recommended procedures. For the statistical analysis, the size of 10 cells per strain per field was determined (6 fields per strain).
In vitro model to test chemicals on ‘Ca. L. asiaticus’ infected leaves
Source of leaves
All leaves used in this study were collected from young flushes that grew on highly symptomatic branches. All shoots were collected from a single HLB-infected, Valencia sweet orange (C. sinensis) tree, maintained by the University of Florida Citrus Research and Education Center (CREC) in Lake Alfred, FL. The infected status of the tree, and widespread distribution of ‘Ca. L. asiaticus’, were confirmed by transmission electron microscopy and PCR analysis of leaf, petiole, and root tissue samples, as described [14], [37]–[39].
Leaf collection and treatment
All solutions were autoclaved or filter sterilized. 100 mM stocks of each chemical were prepared in 100% DMSO. Immediately before collecting leaves, benzbromarone, hexestrol, and phloretin solutions were diluted to 100 µM in ultrapure water. A solution of ultrapure water and DMSO (1%) was used for the controls. A scalpel was used to harvest leaves from the tree, with a horizontal cut at the base of the petiole. Each leaf was immediately suspended in 8 ml of treatment solution (with or without chemicals). Leaves were supported in a vertical position throughout the incubation period, with only the lower inch of the petiole submerged in solution (with or without the chemical). Steady air flow was maintained over the leaf blades throughout the incubation period to facilitate transpiration and the uptake of each solution. Each treatment group consisted of 18 leaves. Nine leaves from each treatment group (including controls) were processed after 6 h of incubation, and the remaining nine leaves were processed after 24 h of incubation.
Leaf tissue processing
For each treatment group, biological triplicates (A, B and C) were prepared from nine leaves. Due to the variable distribution of ‘Ca. Liberibacter asiaticus’ within host trees [37], [40], the tissue from three leaves was combined for each sample. RNA extractions were carried out using the midribs and petioles only. The leaf blades were removed using a scalpel. The remaining midrib and petiole of each leaf was cut into sections (1 cm long) and immediately submerged in RNAlater solution (Life Technologies) as per the manufacturer's instructions. The samples were stored at −80°C until being processed for RNA isolation.
RNA extraction
Plant and bacterial RNA was extracted from midrib and petiole samples using TRI Reagent solution (Sigma-Aldrich), with the addition of a mechanical homogenization step and pressure lysis. Midrib and petiole samples were thawed on ice, and transferred to FT500-S Pulse Tubes (Pressure Biosciences, Easton, MA) with 500 µl of TRI Reagent (Sigma-Aldrich). Samples were homogenized for a total of 2 minutes, in 30 s intervals, on ice, using a PCT Shredder (Pressure Biosciences, Easton, MA). Samples were then transferred to FT500-ND Pulse Tubes (Pressure Biosciences) and subjected to pressure cycling using a NEP 2320 Barocycler (Pressure Biosciences) at 35,000 psi for 30 s, and 0 psi for 30 s, for a total of 20 cycles. Crude lysate was then centrifuged at 5,000× g for 5 min, at 4°C, and the supernatant transferred to a clean RNase free falcon tube for RNA extraction. Chloroform (0.2 volumes) was added to each sample followed by thorough mixing and centrifugation at 5,000× g for 30 min, at 4°C. The aqueous phase was transferred to a clean RNase free falcon tube, and precipitated with isopropanol (0.5 volumes). RNA pellets were washed with 75% ethanol (0.75 vol), briefly air dried, and re-suspended in 100 µl of RNase-free water. RNA samples were treated with RNase-free DNase I for 30 min, at 37°C, followed by DNase Inactivation Reagent (Life Technologies). The concentration of total isolated RNA was determined using a NanoDrop ND 1000 (Thermo Scientific, Wilmington, DE). RNA samples were stored at −80°C. cDNAs were synthesized with M-MLV Reverse Transcriptase (Life Technologies) in accordance with the manufacturer's instructions, using the primers listed in Table 1. Real time quantitative PCR (qRT-PCR) was carried out on as described above. The cox2 gene was measured as an internal plant control. Quantitative reverse transcription-PCR primers are described in detail in Table 1.
Statistical analyses
The statistical significance of data obtained from SEM (cell size) and stress resistance assays (CFU/ml), was determined using a Student's t-test. qRT-PCR statistical significance was assessed using a two-tail P-value, calculated with the Mann–Whitney nonparametric test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ZhangM, Powell Ca, ZhouL, HeZ, StoverE, et al. (2011) Chemical compounds effective against the citrus Huanglongbing bacterium “Candidatus Liberibacter asiaticus” in planta. Phytopathology 101 : 1097–1103.
2. HoffmanMT, DoudMS, WilliamsL, ZhangM-Q, DingF, et al. (2013) Heat treatment eliminates “Candidatus Liberibacter asiaticus” from infected citrus trees under controlled conditions. Phytopathology 103 : 15–22.
3. BishopJL, BoyleEC, FinlayBB (2007) Deception point: peptidoglycan modification as a means of immune evasion. Proceedings of the National Academy of Sciences of the United States of America 104 : 691–692.
4. DavisKM, WeiserJN (2011) Modifications to the peptidoglycan backbone help bacteria to establish infection. Infection and immunity 79 : 562–570.
5. ErbsG, NewmanM (2012) The role of lipopolysaccharide and peptidoglycan, two glycosylated bacterial microbe-associated molecular patterns (MAMPs), in plant innate immunity. Mol Plant Pathol 13 : 95–104 doi:10.1111/J.1364-3703.2011.00730.X
6. BrownPJB, de PedroMA, KyselaDT, Van der HenstC, KimJ, et al. (2012) Polar growth in the Alphaproteobacterial order Rhizobiales. Proceedings of the National Academy of Sciences of the United States of America 109 : 1697–1701.
7. ChauhanS, O'BrianMR (1993) Bradyrhizobium japonicum delta-aminolevulinic acid dehydratase is essential for symbiosis with soybean and contains a novel metal-binding domain. Journal of bacteriology 175 : 7222–7227.
8. CavaF, de PedroMa, LamH, DavisBM, WaldorMK (2011) Distinct pathways for modification of the bacterial cell wall by non-canonical D-amino acids. The EMBO journal 30 : 3442–3453.
9. KovachME, ElzerPH, HillDS, RobertsonGT, FarrisMa, et al. (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166 : 175–176.
10. PagliaiFa, GardnerCL, PandeSG, LorcaGL (2010) LVIS553 transcriptional regulator specifically recognizes novobiocin as an effector molecule. The Journal of biological chemistry 285 : 16921–16930.
11. VedadiM, NiesenFH, Allali-HassaniA, FedorovOY, FinertyPJ, et al. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proceedings of the National Academy of Sciences of the United States of America 103 : 15835–15840.
12. LorcaGL, EzerskyA, LuninVV, WalkerJR, AltamentovaS, et al. (2007) Glyoxylate and pyruvate are antagonistic effectors of the Escherichia coli IclR transcriptional regulator. The Journal of biological chemistry 282 : 16476–16491.
13. LeonardMT, FagenJR, Davis-RichardsonAG, DavisMJ, TriplettEW (2012) Complete genome sequence of Liberibacter crescens BT-1. Standards in genomic sciences 7 : 271–283.
14. FolimonovaSY, AchorDS (2010) Early events of citrus greening (Huanglongbing) disease development at the ultrastructural level. Phytopathology 100 : 949–958.
15. AuclairJL (1963) Aphid Feeding and Nutrition. Annual Review of Entomology 8 : 439–490.
16. Moorby J (1981) Transport Systems in Plants. London; New York: Longman.
17. LavollayM, ArthurM, FourgeaudM, DubostL, MarieA, et al. (2008) The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. Journal of bacteriology 190 : 4360–4366.
18. LamH, OhD, CavaF, TakacsCN, ClardyJ, et al. (2010) D-amino acids govern stationary phase cell wall remodeling in bacteria. NIH Public Access. Science 325 : 1552–1555 doi:10.1126/science.1178123.D-amino
19. BettsJC, LukeyPT, RobbLC, McAdamRa, DuncanK (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Molecular microbiology 43 : 717–731.
20. YanQ, SreedharanA, WeiS, WangJ, Pelz-StelinskiK, et al. (2013) Global gene expression changes in Candidatus Liberibacter asiaticus during the transmission in distinct hosts between plant and insect. Molecular plant pathology 14 : 391–404.
21. BaumgarthB, BartelsFW, AnselmettiD, BeckerA, RosR (2005) Detailed studies of the binding mechanism of the Sinorhizobium meliloti transcriptional activator ExpG to DNA. Microbiology 151 : 259–268.
22. ZhuD, WangY, ZhangM, IkedaH, DengZ, et al. (2013) Product-mediated regulation of pentalenolactone biosynthesis in Streptomyces species by the MarR/SlyA family activators PenR and PntR. Journal of bacteriology 195 : 1255–1266.
23. BartelsFW, BaumgarthB, AnselmettiD, RosR, BeckerA (2003) Specific binding of the regulatory protein ExpG to promoter regions of the galactoglucan biosynthesis gene cluster of Sinorhizobium meliloti - a combined molecular biology and force spectroscopy investigation. Journal of Structural Biology 143 : 145–152.
24. HammesW, SchleiferKH, KandlerO (1973) Mode of action of glycine on the biosynthesis of peptidoglycan. Journal of bacteriology 116 : 1029–1053.
25. VanderlindeEM, YostCK (2012) Mutation of the sensor kinase chvG in Rhizobium leguminosarum negatively impacts cellular metabolism, outer membrane stability, and symbiosis. Journal of bacteriology 194 : 768–777.
26. SkinnerFA (1977) An Evaluation of the Nile Blue Test for Differentiating Rhizobia from Agrobacteria. Journal of Applied Bacteriology 43 : 91–98.
27. DubéeV, TribouletS, MainardiJ-L, Ethève-QuelquejeuM, GutmannL, et al. (2012) Inactivation of Mycobacterium tuberculosis l,d-transpeptidase LdtMt1 by carbapenems and cephalosporins. Antimicrobial agents and chemotherapy 56 : 4189–4195.
28. SambrookJ (2008) Molecular Cloning.
29. NiesenFH, BerglundH, VedadiM (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols 2 : 2212–2221.
30. ZianniM, TessanneK, MerighiM, LagunaR, TabitaFR (2006) Ab r f. 17 : 103–113.
31. Guérout-FleuryaM, FrandsenN, StragierP (1996) Plasmids for ectopic integration in Bacillus subtilis. Gene 180 : 57–61.
32. Petit-GlatronMF, ChambertR (1992) Peptide carrier potentiality of Bacillus subtilis levansucrase. Journal of general microbiology 138 : 1089–1095.
33. Miller JH (1972) Experiments in Molecular Genetics. Cold Springs Harbor (New York): Cold Spring Harbor Laboratory Press.
34. GaoM, ChenH, EberhardA, GronquistR, RobinsonJB, et al. (2005) sinI - and expR -Dependent Quorum Sensing in Sinorhizobium meliloti sinI - and expR-Dependent Quorum Sensing in Sinorhizobium meliloti †. J Bacteriol 187 : 7931–7944 doi:10.1128/JB.187.23.7931
35. OkeV, LongSR (1999) Bacterial genes induced within the nodule during the Rhizobium-legume symbiosis. Molecular microbiology 32 : 837–849.
36. FinanTM, KunkelB, De VosGF, SignerER (1986) Second symbiotic megaplasmid in Rhizobium meliloti carrying exopolysaccharide and thiamine synthesis genes. Journal of bacteriology 167 : 66–72.
37. HartungJS, PaulC, AchorD, BrlanskyRH (2010) Colonization of dodder, Cuscuta indecora, by “Candidatus Liberibacter asiaticus” and “Ca. L. americanus”. Phytopathology 100 : 756–762.
38. LiW, HartungJS, LevyL (2006) Quantitative real-time PCR for detection and identification of Candidatus Liberibacter species associated with citrus huanglongbing. Journal of microbiological methods 66 : 104–115.
39. HilfME, SimsKR, FolimonovaSY, AchorDS (2013) Visualization of “Candidatus Liberibacter asiaticus” cells in the vascular bundle of citrus seed coats with fluorescence in situ hybridization and transmission electron microscopy. Phytopathology 103 : 545–554.
40. Pelz-StelinskiKS, BrlanskyRH, EbertTa, RogersME (2010) Transmission Parameters for Candidatus Liberibacter asiaticus by Asian Citrus Psyllid (Hemiptera: Psyllidae). Journal of Economic Entomology 103 : 1531–1541.
41. GalibertF, FinanTM, LongSR, PuhlerA, AbolaP, et al. (2001) The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293 : 668–672 Available: http://www.ncbi.nlm.nih.gov/pubmed/11474104. Accessed 24 January 2014.
42. GaoM, ChenH, EberhardA, GronquistMR, RobinsonJB, et al. (2005) sinI - and expR-dependent quorum sensing in Sinorhizobium meliloti. Journal of bacteriology 187 : 7931–7944.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus