
Zinc Finger Independent Genome-Wide Binding of Sp2 Potentiates Recruitment of Histone-Fold Protein Nf-y Distinguishing It from Sp1 and Sp3
A major question in eukaryotic gene regulation is how transcription factors with similar structural features elicit specific biological responses. We used the three transcription factors Sp1, Sp2 and Sp3 as a paradigm for investigating this question. All three proteins are ubiquitously expressed, and they share glutamine-rich domains as well as a conserved bona fide zinc finger DNA binding domain. Yet, each of the three proteins carries out unique functions in vivo, and each is absolutely essential for mouse development. By genome-wide binding analysis, we found that Sp1 and Sp3 on the one hand, and Sp2 on the other hand engage completely different protein domains for their genomic binding site selection. Most strikingly, the zinc finger domain of Sp2 is dispensable for recruitment to its target sites in vivo. Moreover, we provide strong evidence that the histone-fold protein Nf-y is necessary for recruitment of Sp2. Conversely, Sp2 potentiates Nf-y binding showing that binding of Sp2 and Nf-y to shared sites is mutually dependent. Our findings uncover an unexpected mechanistic diversity in promoter recognition by seemingly similar transcription factors. This work has broader implications for our understanding of how members of other multi-protein transcription factor families could achieve specificity.
Published in the journal:
Zinc Finger Independent Genome-Wide Binding of Sp2 Potentiates Recruitment of Histone-Fold Protein Nf-y Distinguishing It from Sp1 and Sp3. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005102
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005102
Summary
A major question in eukaryotic gene regulation is how transcription factors with similar structural features elicit specific biological responses. We used the three transcription factors Sp1, Sp2 and Sp3 as a paradigm for investigating this question. All three proteins are ubiquitously expressed, and they share glutamine-rich domains as well as a conserved bona fide zinc finger DNA binding domain. Yet, each of the three proteins carries out unique functions in vivo, and each is absolutely essential for mouse development. By genome-wide binding analysis, we found that Sp1 and Sp3 on the one hand, and Sp2 on the other hand engage completely different protein domains for their genomic binding site selection. Most strikingly, the zinc finger domain of Sp2 is dispensable for recruitment to its target sites in vivo. Moreover, we provide strong evidence that the histone-fold protein Nf-y is necessary for recruitment of Sp2. Conversely, Sp2 potentiates Nf-y binding showing that binding of Sp2 and Nf-y to shared sites is mutually dependent. Our findings uncover an unexpected mechanistic diversity in promoter recognition by seemingly similar transcription factors. This work has broader implications for our understanding of how members of other multi-protein transcription factor families could achieve specificity.
Introduction
Eukaryotic transcription factors are grouped into families based on their common structural features. Prototypical zinc finger-containing transcription factors are the evolutionary conserved Specificity proteins/Krüppel-like factors (Sps/Klfs) (reviewed in [1,2,3,4]) that share three consecutive C2H2-type zinc fingers in their C-terminal moiety. Mammals have nine different Sp factors (Sp1 to Sp9), which can be grouped into two subclasses based on structural features outside of the zinc finger domain [5]. The Sp1 to Sp4 subclass is characterized by glutamine-rich domains that have been shown to act as transactivation domains in Sp1, Sp3 and Sp4. Sp1, Sp2 and Sp3 are ubiquitously expressed whereas expression of Sp4 is largely restricted to neuronal cells [1,6].
Despite their structural similarities and broad co-expression there seems to be little functional overlap between Sp1, Sp2 and Sp3 [7,8,9]. Briefly, Sp1null as well as Sp2null embryos are severely growth-retarded and die before embryonic day 10 [7,9]. Conditional inactivation of Sp2 in neuronal stem cells and neuronal progenitor cells resulted in impaired proliferation and disrupted neurogenesis in embryonic and postnatal brain [10]. In homozygous Sp2 transgenic mice terminally differentiated keratinocytes are depleted and the animals die within two weeks after birth again underlining the physiological importance of Sp2 [11]. Moreover, a genome-wide screen for cell division genes identified Sp2 as a gene essential for proper mitosis in HeLa cells [12]. Finally, Sp3null mice develop until birth but are not viable due to manifold defects including impaired lung, cardiac, bone and red blood cell development [8,13,14,15].
At the molecular level, the functional properties of Sp1 and Sp3 are well characterized. Particularly, numerous publications reported binding of Sp1 and Sp3 to the GC box (GGGGCGGGG) and related motifs in vitro. In contrast, Sp2 has largely escaped attention since its initial discovery [16] likely because no DNA-binding activity of full-length Sp2 is detectable by the electrophoretic mobility shift assay [17,18], and because Sp2 has little activation capacity on promoters that are regulated by Sp1 or Sp3 in reporter gene assays [19].
We have recently determined the genome-wide occupancy of Sp2 in mouse embryonic fibroblasts (MEFs) and in HEK293 cells, and have found that Sp2 occupies numerous proximal promoters of essential genes [18]. Bioinformatics analysis of the Sp2 binding sites identified CCAAT - and GC boxes as prevalent motifs in these promoters. On this basis and taking into account the similarity of Sp2 with other Sp factors, we concluded that Sp2 is recruited to its sites in chromatin by binding to the GC box in vivo. However, in the global chromatin context, it remains a largely unanswered question whether the very similar transcription factors Sp1, Sp2 and Sp3 are bound to the same promoters in vivo and whether regions outside the bona fide DNA-binding domain contribute to their binding site selection.
To address this important question, we have compared the genome-wide chromatin occupancy of Sp1, Sp2 and Sp3 with each other in mouse embryonic fibroblasts. We found that Sp1 and Sp3 essentially occupy the same promoters and localize to GC boxes. In marked contrast, Sp2 predominantly localizes at CCAAT motifs. By re-expression of various Sp2 and Sp3 mutants in corresponding Sp2 and Sp3 knockout (Sp2ko and Sp3ko) MEFs, we found that the zinc finger region mediates chromatin binding of Sp3. Unexpectedly, the bona fide zinc finger DNA-binding domain is completely dispensable for binding of Sp2. Rather, it is exclusively the glutamine-rich N-terminal domain, which mediates recruitment of Sp2 to its genomic sites. We further show that Sp2 colocalizes with the trimeric CCAAT-binding transcription factor Nf-y at a large fraction of Sp2 binding sites. We provide evidence that Nf-y is necessary for recruitment of Sp2 and suggest that, in turn, Sp2 potentiates Nf-y binding to shared sites, since binding of Nf-y to sites that are also bound by Sp2 is attenuated in Sp2ko MEFs. Therefore, we have discovered that the seemingly similar transcription factors Sp1/Sp3 and Sp2 utilize completely different modes of genomic binding site selection and shed light on the previously enigmatic properties of Sp2.
Results
ChIP-seq analysis indicate that Sp1 and Sp3 occupy the same promoters
Recently, we have identified genomic binding sites of the transcription factor Sp2 in MEFs [18]. To identify the binding sites for the related transcription factors Sp1 and Sp3, and to elucidate the potential overlap with Sp2, we performed ChIP-seq analysis with the same cells. Two different antibodies for each factor that do not cross-react with other Sp family members were used ([8,18] and S1–S2 Figs). An Sp3 ChIP using Sp3ko MEFs served as a control for the selection of Sp3-specific peaks; and an IgG ChIP for the selection of Sp1-specific peaks, as Sp1ko MEFs are not viable.
We identified 5589 Sp1 and 4041 Sp3 peaks as overlapping across two sets of samples using two different antibodies for each factor (Fig. 1A). Comparing the Sp1 and Sp3 peaks of all four ChIP-seq data sets revealed 3597 high-confidence sites that are bound by Sp1 as well as by Sp3 (Fig. 1B). The large majority of these sites (~93%) are located close to the 5´-end (+/ - 500 bp) of annotated transcripts (Fig. 1C). The comparison of the Sp1 - and Sp3 ChIP-seq data sets also revealed a fraction of ~700 Sp1-specific peaks and a few (81) Sp3-specific peaks (Fig. 1B). The majority of these sites represent peaks with relatively low tag counts just above the threshold used for peak selection. Another fraction of the potential Sp1-specific peaks is also found in the Sp3 ChIP-seq data sets. However, these peaks were removed from the classified Sp3 list because a peak appeared also in Sp3ko MEFs. The latter observation highlights the great benefit of using knockout cells as ChIP controls. Taken together, we are not convinced about the reliability of the apparently specific Sp1 - or Sp3 binding sites. Rather, our ChIP-seq results support the notion that Sp1 and Sp3 essentially occupy the same promoters in vivo. Combined binding of Sp1 and Sp3 to the same promoters is consistent with the phenotype of Sp1/Sp3 compound heterozygous mice. These mice are not viable suggesting that a critical threshold of Sp1 and Sp3 activity is required for normal embryonic development and for proper regulation of common target genes [20].

Genomic binding of Sp1/Sp3 and Sp2 is different
We next compared the Sp1/Sp3 binding sites with those of Sp2 [18]. Although there is a large overlap, we were able to distinguish, with high confidence, Sp1/Sp3-specific as well as Sp2-specific binding sites (Fig. 1D). Genome browser snapshots of representative shared Sp1/Sp2/Sp3, Sp1/Sp3-specific, and Sp2-specific binding sites are shown in Figs. 1E and S2. We also probed a panel of selected target promoters by conventional ChIP-qPCR analysis, and confirmed binding of all three Sp-factors to common promoters such as the L3mbtl2, Nxt1, Bin3 and Dhfr promoter, Sp1/Sp3-specific binding to the Raf1, Calcoco1, Kdelr2 and the Grb2 upstream promoter, and Sp2-specific binding to the Oxr1, Plcl1, and the Nfyc and Grb2 downstream promoters (Fig. 1F), the latter containing alternative promoters that are either occupied by Sp1/Sp3 or Sp2. Of note, consistent with our previous observation [18], Sp1 and Sp3 binding to several promoters is reduced in Sp2ko MEFs, which is likely due to their lower expression levels in these cells [18].
Sp1/Sp3 and Sp2 binding sites show different sequence motif distributions
We performed an unsupervised de novo sequence motif analysis at Sp1/Sp3 and Sp2 binding sites. The top motif at the Sp1/Sp3 peaks matches well-known in vitro Sp1/Sp3 binding sites with the GGGCGGG core sequence (GC box). The second enriched motif is “CCAAT”, which is a binding site for the transcription factor Nf-y. Essentially, the same two motifs are found at the Sp2 binding sites [18]. However, at the Sp2 binding sites, the CCAAT motif is much more prevalent than the GC box motif (Fig. 2A). Moreover, only the GC box motif but not the CCAAT motif is enriched at sites that are bound by Sp1 and Sp3 but not by Sp2. Conversely, only the CCAAT motif but not the GC box is enriched at sites that are bound by Sp2 but not by Sp1 and Sp3 (Fig. 2A). To extract positional information for the GC and CCAAT motifs within Sp1/Sp3 and Sp2 peaks, we performed a central motif enrichment analysis (CMEA) [21]. This analysis revealed that the GC box motif is enriched at the peak centers of the Sp1/Sp3 binding sites, whereas the CCAAT box motif is found at flanking regions showing a multimodal shape (Fig. 2B). A strikingly different picture emerges at the Sp2 binding sites. Most significantly, the GC box motif is barely enriched at the Sp2 peak centers, whereas the CCAAT box motif exhibits a centrally enriched, symmetrical bimodal distribution with a mean distance of ~35 bp (Fig. 2B).
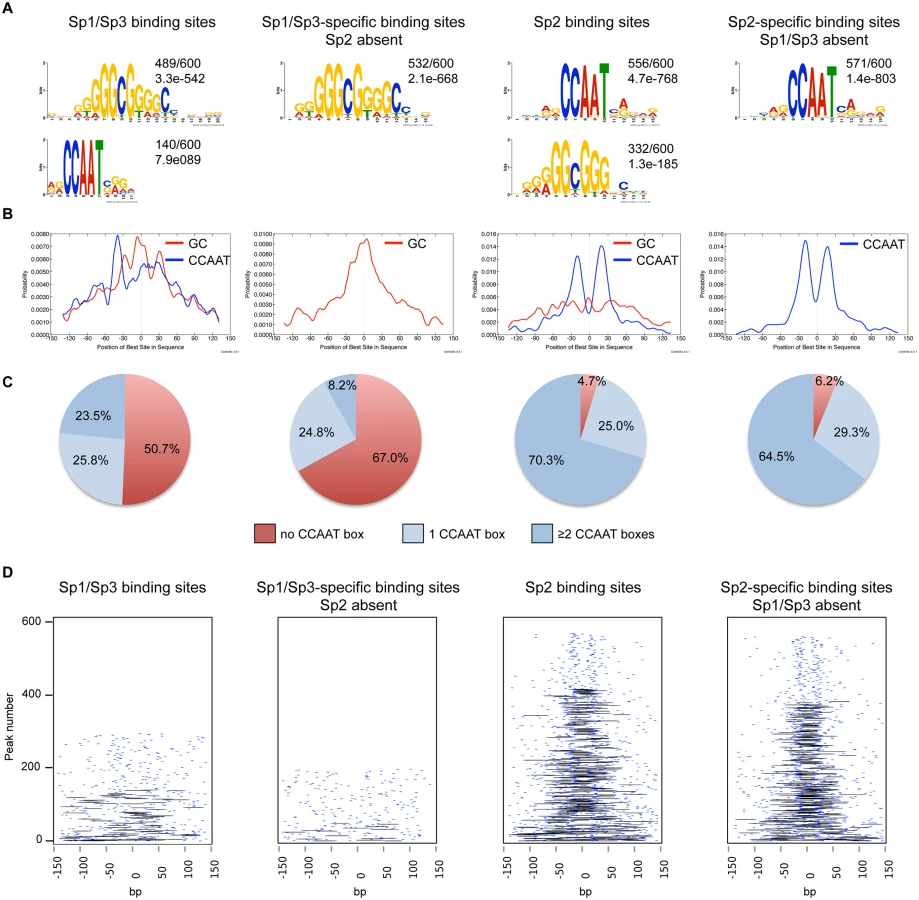
Intrigued by the nicely shaped distribution of the CCAAT motifs at the Sp2 binding sites, we examined the distribution of the CCAAT motif in greater detail. We found that ~70% of the top Sp2 binding sites but only ~23% of the Sp1/Sp3 sites contain two, or more, perfect CCAAT motifs. Moreover, ~65% of the Sp2-specific sites but only ~8% of the Sp1/Sp3-specific sites contain more than one CCAAT motif (Fig. 2C). Finally, many Sp2-specific promoters (~40%) but only a few Sp1/3-specific promoters (2.3%) contain two CCAAT/ATTGG motifs that are located within a distance of 30 to 50 nucleotides (Fig. 2D). Thus it appears that a large fraction of the Sp2 binding sites is characterized by tandem CCAAT motifs. The CCAAT motif is a binding site for the transcription factor Nf-y. Since Nf-y is also found at promoters that contain imperfect CCAAT motifs [22], the number of Sp2 binding sites with tandem arranged CCAAT motifs (e.g. Nf-y binding sites, see below) could be even higher.
Finally, we also determined the binding sites of Sp1 in HEK293 cells and compared the motifs at the Sp1 binding sites with those at the Sp2 binding sites [18]. Similar to mouse cells, the GC box is the prevalent motif at the Sp1 binding sites, whereas the CCAAT box is the prevalent motif at the Sp2 binding sites (S3 Fig). In summary, the comparison of the genomic Sp1/Sp3 and Sp2 binding sites revealed markedly different sequence motif distributions indicating that binding of Sp2 to chromatin is distinct from Sp1/Sp3, and questions our previous conclusion that Sp2 is recruited to its target promoters in vivo via binding to the GC box [18].
The Sp2 DNA-binding domain is dispensable for genomic binding
Intrigued by the finding that Sp1 and Sp3 are present at GC boxes, whereas Sp2 is located primarily at CCAAT motifs, we sought to identify the protein domains that are responsible for the differences in binding site selection in vivo. We focused on Sp2 and Sp3 because corresponding knockout MEFs are available [7,23] that have been proven to be useful for rescue experiments [18,24,25].
We stably expressed Flag-tagged full-length Sp2 and Sp3, and deletion mutants thereof in corresponding knockout MEFs. All proteins are expressed at levels similar to endogenous Sp2 and Sp3 with the exception of the zinc finger domains that are expressed at a higher level (Fig. 3A-B). ChIP-qPCR analysis of selected target promoters including promoters that are bound by Sp2 as well as by Sp3 (Nxt1, Sp1 and Sp2), and promoters that are preferentially bound by either Sp2 (Gas2l3 and Osbp) or Sp3 (Calcoco1 and Raf1) revealed specific binding of re-expressed full-length Sp2 and the full-length Sp3 isoforms (Sp3li and Sp3si) (Fig. 3C-D). The Sp3ZF fragment lacking the entire N-terminal part is also bound at the Sp3 target promoters showing that the Sp3 zinc finger domain is sufficient for binding. As expected, the Sp3NT mutant lacking the DNA-binding domain is not bound at any of these promoters (Fig. 3D). The picture, which emerged from the analysis of the Sp2 mutants is completely different. The Sp2ZF fragment lacking the N-terminal part is not bound at any of the Sp2 target promoters (Fig. 3C). Strikingly, the Sp2NT mutant expressing the entire N-terminal part but lacking all three zinc fingers is bound at the Nxt1, Sp1, Sp2, Gas2l3 and Osbp promoters almost as strongly as full-length Sp2 (Fig. 3C). Thus, the canonical DNA-binding domain of Sp2 is dispensable, and the N-terminal part sufficient for binding to these promoters in vivo. This result suggests that Sp2 is recruited to its target promoters indirectly, likely involving protein-protein interactions (see Discussion chapter for further details).

To further substantiate the conclusion that Sp2 is recruited to its genomic sites by the N-terminal region rather than by the C-terminal zinc finger domain, we performed ChIP-seq using Sp2ko MEFs re-expressing Flag-tagged versions of full-length Sp2 (Sp2FL), the Sp2 N-terminal part (Sp2NT) or the Sp2 zinc finger domain (Sp2ZF; see scheme in Fig. 3A). Although the Flag ChIP was less efficient in this particular experiment, we identified more than seven hundred highly reliable Sp2FL and Sp2NT binding sites but only ten potential Sp2ZF sites (Fig. 4A). Comparison of these binding sites with those of endogenous Sp2 in wild type MEFs revealed that >99% of the Sp2FL and >95% of the Sp2NT sites, but remarkably none of the Sp2ZF sites correspond to native Sp2 binding sites (Fig. 4A). The correlation of normalized tag counts at individual Flag-Sp2FL and Flag-Sp2NT peaks (Fig. 4B) shows that full-length Sp2 and the zinc finger-deficient Sp2NT mutant bind to chromatin with similar strength. Of note, the sites bound by the Sp2NT moiety correspond to the strongest native Sp2 peaks (Fig. 4C). Representative genome browser snapshots of Sp2FL and Sp2NT peaks in comparison with native Sp2 peaks are shown in Fig. 4D. Taken together, these striking results show convincingly that the zinc finger domain is dispensable for genomic binding of Sp2 to its target promoters. Recruitment of Sp2 in vivo by its N-terminal region is not limited to just a few selected target sites, but is a general feature of Sp2-targeting to chromatin (Fig. 4E).

To map protein regions of Sp2 that are essential for chromatin binding in vivo, we stably re-expressed a series of N-terminal Sp2 deletion mutants (Fig. 5A) in Sp2ko MEFs and tested their binding to selected loci. Western blotting and immunohistochemistry control experiments showed that all mutants are expressed at similar levels and are present in the nucleus (Fig. 5B-C). Deletion of 27 N-terminal amino acids does not affect binding of Sp2, whereas deletion of 93 N-terminal amino acids completely abolishes binding (Fig. 5D). The Sp2 mutant lacking 49 N-terminal amino acids shows some residual binding to the Sp2 and Osbp promoters. We conclude that the Sp2 sequence between amino acids 28 and 93 is essential for binding of Sp2 in vivo. This region contains the Sp-box (33-SPLALLAATCSKIG-46), a hallmark of the Sp transcription factor family members [1]. Whether the Sp-box is directly involved in recruitment of Sp2 remains to be established. Finally, we also rescued Sp2ko MEFs with a C-terminal deletion mutant (1–506 mutant in Fig. 5A) that lacks the zinc finger domain and, in addition, the buttonhead box. The buttonhead box is a cysteine-rich motif (CxCPnC) of unknown function, and it is also a hallmark of Sp factors. The Sp2 1–506 mutant binds to the Sp2, Osbp and Amd1 promoters as efficiently as full-length Sp2 demonstrating that the buttonhead box is also dispensable for binding of Sp2 to its sites in vivo.

The N-terminal region of Sp2 rescues target gene expression
We wanted to know whether binding of the Sp2NT fragment could affect expression of target genes. At first, we tested whether it has the capacity to activate transcription. We fused the Sp2NT fragment to the Gal4-DNA-binding domain and performed reporter gene assays. The Gal4-Sp2NT fusion protein activated a Gal4-responsive 5xUAS-luciferase reporter as efficiently as a corresponding Gal4-Sp1NT fusion protein (Fig. 6A) showing that the N-terminal part of Sp2 has an activation function.

Next, we tested expression of a selection of Sp2 target genes in wild type, Sp2ko, and Sp2ko MEFs re-expressing Sp2 mutants. Compared to wild type MEFs, Nlk transcript levels are markedly lower in Sp2ko MEFs (20% of wt). Nearly wild type Nlk mRNA levels are restored in Sp2ko MEFs expressing either full-length Sp2 or the Sp2NT mutant, but not in cells expressing the Sp2ZF mutant (Fig. 6B). This result indicates that the loss of Sp2 leads to downregulation of Nlk transcription, and that re-expression of the Sp2 N-terminal fragment is sufficient to rescue Nlk expression. Nevertheless, the expression levels of many other Sp2 target genes were not significantly affected in Sp2ko MEFs [18]. Inspection of annotated ensemble transcripts revealed that many Sp2 target genes (65%) have alternative transcriptional start sites. Moreover, many of the Sp2 binding sites overlap with Sp1/Sp3 binding sites (Fig. 1). Therefore, we reasoned that alternative initiation sites might hide transcriptional initiation driven by Sp2. We chose the Grb2 and the Oxr1 genes to test this possibility. Both genes contain an upstream promoter bound by Sp1 and Sp3 but not by Sp2 and a downstream promoter bound by Sp2 but not by Sp1 and Sp3 (Fig. 6C). We analyzed levels of Grb2 and Oxr1 transcripts with primer pairs that detect all transcripts, and with primer pairs that detect specifically the transcripts initiated at the Sp2-specific downstream promoters. Compared to wild type MEFs, the total transcript levels of the Grb2 and Oxr1 genes are largely unaffected in Sp2ko MEFs. In contrast, the downstream promoter-specific transcript levels are significantly lower in Sp2ko MEFs; and they are at least partially restored in Sp2ko MEFs expressing full-length Sp2 or the Sp2NT mutant (Fig. 6C). In summary, these results indicate that the N-terminal region of Sp2 is sufficient for activation of a subset of genes. However, they do not exclude that the zinc finger domain of Sp2 is essential for activation of other genes.
The majority of Sp2 sites are also bound by Nf-y
The CCAAT box is the binding site of the ubiquitous transcription factor nuclear factor y (Nf-y, previously also termed Cbf for CCAAT-binding factor; reviewed in [26]). Nf-y is a heterotrimeric protein composed of the three subunits Nf-ya, Nf-yb and Nf-yc. The Nf-ya subunit confers base-specific recognition of the CCAAT sequence, whereas the basic surface of the histone-fold Nf-yb/Nf-yc dimer makes strong contacts with the negatively charged DNA sugar-phosphate backbone [27]. All three Nf-y subunits are necessary for binding to the CCAAT box. Based on its histone-like properties, Nf-y is considered as an architectural promoter organizer that keeps a promoter free of nucleosomes [27].
The presence of Sp2 at CCAAT boxes rather than at the GC box prompted us to ask whether Nf-y is also present at these sites, and whether genomic binding of Nf-y and Sp2 impinge on each other. We performed ChIP-seq of Nf-ya, Nf-yb and Nf-yc with chromatin from wild type and Sp2ko MEFs (Fig. 7A). We obtained a higher number of Nf-yb sites with respect to Nf-ya and Nf-yc sites, which very likely does not reflect the occurrence of Nf-yb-specific target loci, but a better ChIP efficiency of the Nf-yb antibody. This assumption is supported by the significantly higher average reads per peak obtained for Nf-yb as well as by the higher enrichment values obtained in ChIP-qPCR experiments (see below). Comparison of the Sp2 and the Nf-y ChIP-seq data sets revealed that 84% of the Sp2 target sites are also bound by Nf-y (Fig. 7B). Moreover, the strength of Nf-y - and Sp2 binding correlates with each other; in other words, sites with high Nf-y subunit tag counts have also high Sp2 tag counts (Fig. 7C). Nevertheless, a subset of sites is clearly bound exclusively by Sp2 or exclusively by Nf-y. Examples of such sites are the Fanci and the Taf1c promoters that are bound by Sp2 but not by Nf-y, and the Atxn3 and the Wapal promoters that are bound by Nf-y but not by Sp2 (Fig. 7D).

We compared sites that are bound by Sp2 as well as by Nf-y with sites that are bound only by Sp2 or only by Nf-y. MEME reported the CCAAT and the GC motif as the top motifs at sites that are co-bound by Sp2 and Nf-y as well as at sites that are bound by Nf-y but not by Sp2 (Fig. 7E). Consistent with the analysis of all Sp2 ChIP-seq peaks, the CCAAT motif shows a symmetric bimodal distribution at shared Sp2/Nf-y sites. The CCAAT motif at the Nf-y-specific sites does not show this pronounced bimodal distribution but is largely centrally enriched (Fig. 7F). The prevalent motifs at sites bound by Sp2 but not by Nf-y are the GGAAG motif, a binding site for the Ets family members Gabp and Elk4, and the GC box (Fig. 7E). At these sites, the GC box appears to be centrally enriched and flanked by the GGAAG motif (Fig. 7F).
The occurrence of a centrally enriched GC box made it possible that the zinc finger domain of Sp2 could be essential for binding to these sites. Notably, these sites are weak binding sites (S4 Fig) that were not detected in our Flag-Sp2FL and Flag-Sp2NT ChIP-seq experiments shown in Fig. 4. Therefore, we tested binding of Sp2 mutants to this class of promoters by conventional ChIP-qPCR. Full-length Sp2 as well as the Sp2NT fragment are bound to the Fanci and the Taf1c promoters (S4 Fig). We conclude that the zinc finger region of Sp2 is also dispensable for binding to sites that are not bound by Nf-y.
Finally, we determined the overlap of Sp1/Sp3, Sp2 and Nf-y sites. Approximately 45% of the Sp1/Sp3 sites are also occupied by Nf-y (Fig. 7G). Importantly, the vast majority of the Sp1/Sp3 binding sites that are not bound by Nf-y are also not bound by Sp2 (1825 of 1967; 93%) (Fig. 7G). These sites represent high tag count Sp1/Sp3 binding sites that are enriched of multiple GC boxes and, as expected, do not contain CCAAT motifs (Fig. 7H). Thus, the occurrence of GC boxes and the presence of Sp1/Sp3 are not sufficient to recruit Sp2. Of note, the prevalent, centrally enriched motif at the few Sp2 binding sites that are also bound by Sp1/3 but not by Nf-y (142 sites in Fig. 7G) is the GGAAG motif and not the GC box (Fig. 7H). These findings have also implications concerning the binding of Sp2 to promoters that contain both, GC and CCAAT boxes and are bound by Sp1/Sp3 and by Nf-y (Figs. 2, 7G). At these promoters, the resolution of the ChIP-seq peaks does in most cases not allow to distinguish whether Sp2 is located at a GC box or at a nearby CCAAT box. Given that Sp2 is bound to a large fraction of promoters that are bound by Nf-y lacking Sp1/Sp3 (1081 sites; 42%), it is tempting to conclude that the CCAAT box (i.e. Nf-y) and not the GC box (i.e. Sp1/Sp3) is the decisive sequence for the recruitment of Sp2 to promoters bound by Nf-y and by Sp1/Sp3.
Sp2 and Nf-y bind simultaneously to shared sites
Having established the overlap of Sp2 and Nf-y target sites, we tested whether Sp2 and Nf-y simultaneously associate with chromatin, and performed sequential ChIP experiments (re-ChIP). The eluate from Sp2 antibody chromatin precipitation was subjected to precipitation with Nf-yb antibodies, and vice versa. Re-ChIPs detected all selected promoters bound by Sp2 and Nf-y (Sp1, Sp2, Osbp, Amd1, Nxt1 and Nipal3) independently of the immunoprecipitation order (Fig. 8A), but not the Sp2-specific Fanci, the Nf-y-specific Atxn3 or the Sp1/Sp3-specific Raf1 promoter. This result demonstrates that Sp2 and Nf-y co-occupy their shared binding sites in the context of chromatin.

Nf-y is necessary for Sp2 binding
Co-occupancy of Sp2 and Nf-y at shared target promoters led us to ask whether Nf-y is necessary for recruitment of Sp2 to these sites. We knocked down all three Nf-y subunits individually by RNAi (Fig. 8B), and subsequently analyzed binding of Nf-y and Sp2 to a panel of target promoters. These promoters include those that are co-bound by Nf-y and all three Sp factors (Sp1, Osbp, Amd1 and Dcnt4), promoters that are co-bound by Nf-y and Sp2 but not by Sp1 and Sp3 (Oxr1 and Plcl1), and promoters that are bound by all three Sp factors but not by Nf-y (Fanci and Taf1c) (Fig. 8C). Lower expression of any of the Nf-y subunits resulted in attenuated binding of Sp2 to all promoters that are co-bound by Nf-y and Sp2. Importantly, the reduction of Sp2 binding is particularly strong at the Plcl1 and Oxr1 promoters, which are not bound by Sp1 and Sp3. This result strongly suggests that reduced binding of Sp2 is not an indirect effect of reduced Sp1 levels in Nf-ya and Nf-yc depleted cells (Fig. 8B, panel 4, lanes 2 and 4) but directly caused by attenuated Nf-y binding. Binding of Sp2 to the Fanci and Taf1c promoters, which are not co-bound by Nf-y, was not affected upon depletion of Nf-yb and Nf-yc. Reduced binding of Sp2 to these two promoters upon Nf-ya knockdown is likely due to lower Sp2 levels in Nf-ya knockdown cells (see Fig. 8B, panel 3, lane 2). Taken together, these results strongly suggest that the presence of Nf-y is necessary for recruitment of Sp2 to shared sites.
Sp2 potentiates binding of Nf-y
Given the dependency of Sp2 on Nf-y, we wanted to know whether Sp2 does affect binding of Nf-y at these shared sites. We compared occupancy of Nf-y in wild type MEFs with occupancy in Sp2ko MEFs. Expression of Nf-y is similar in both cell types (Fig. 9A). By calculating the ratio of normalized Nf-y ChIP-seq tag counts in wt and in Sp2ko MEFs (wt/Sp2ko), we found attenuated Nf-y occupancy in Sp2ko MEFs at many promoters that are co-bound by Sp2 in wild type MEFs (Fig. 9B, top panel). Attenuated Nf-y binding in Sp2ko cells is particularly strong at promoters that are bound in wild type MEFs by Sp2 but not by Sp1/3 such as the Nrxn2, Grb2, Nipal3, Plcl1 or the Oxr1 promoters (Figs. 9C-D, S5). This demonstrates that binding of Nf-y to these promoters depends on the presence of Sp2. Importantly, Nf-y binding remains largely unchanged or is even slightly potentiated at sites that are not co-bound by Sp2 (Fig. 9B, bottom panel) including promoters that are also bound by Sp1/Sp3 such as the Atxn3, Mxi1, and Pdcd4 promoters (Figs. 9C-D, S6).

Given that genomic Nf-y binding is attenuated in Sp2ko MEFs at loci that are co-bound by Sp2, it is to be expected that re-expression of Sp2 potentiates Nf-y binding at these sites. Indeed, we found stronger Nf-y binding in rescued MEFs, specifically at loci that are re-occupied by Sp2FL or by Sp2NT (Sp2, Osbp, Amd1, Nipal3 and Nlk) (Fig. 9E). Binding of Nf-y at these promoters remained unchanged in cells expressing the Sp2ZF fragment. Finally, binding of Nf-y to the Atnx3 promoter, which is not an Sp2 target, is similar in Sp2FL, Sp2NT and Sp2ZF expressing cells (Fig. 9E). Taken together, these results strongly support the conclusion that Sp2 potentiates binding of Nf-y at shared sites.
Discussion
In this study, we have elucidated the mode of genomic binding site selection of the zinc finger transcription factors Sp1, Sp2 and Sp3. A key finding is that Sp1/3 and Sp2 bind to chromatin by distinct mechanisms. Consistent with numerous in vitro DNA-binding studies, Sp1 and Sp3 localize to GC boxes and essentially occupy the same promoters. Unexpectedly, Sp2 primarily localizes at CCAAT motifs, often arranged in tandem with a mean distance of 35 bp (Fig. 2). In line with their different binding site selection, re-expression of Sp2 and Sp3 mutants in corresponding Sp2ko and Sp3ko MEFs revealed that different protein domains mediate binding to their sites in chromatin. As expected, the C-terminal zinc finger domain is essential and largely sufficient for binding of Sp3 to its target promoters. Although not formally tested, we consider it as very likely that the zinc finger domain of Sp1 also mediates binding to chromatin. Intriguingly, the zinc finger domain of Sp2 is fully dispensable for binding in vivo. Instead, binding of Sp2 to its target promoters is mediated by the N-terminal glutamine-rich part, providing the essential clue for the different binding site selection of Sp2, as compared to Sp1 and Sp3.
Interestingly, Sp2 targeted mice that do not express the zinc finger domain but aberrantly express an RNA encoding the N-terminal region display a markedly less severe phenotype than Sp2null mice [7]. Although expression remains to be established at the protein level, binding of the Sp2 N-terminal region to chromatin shown in this report provides a rational explanation for the different phenotypes of these mice.
Sequence comparisons of the N-terminal region of Sp2 with the corresponding regions of Sp1 and Sp3 reveal major differences of their amino acid composition. Most significantly, in addition to the frequent occurrence of glutamine residues, the N-terminal domains of Sp1 and Sp3 are rich in acidic amino acids, whereas the N-terminal domain of Sp2 is very rich in basic amino acids (S7 Fig). Thus, the general assignment of the N-terminal domains of Sp1, Sp2 and Sp3 as glutamine-rich domains is misleading. Importantly, the presence of multiple positive charged lysines in the N-terminal region of Sp2 raises the possibility of non-specific interactions with the negatively charged sugar-phosphate backbone of the DNA.
Binding sites of Sp1 and Sp2 have also been identified in K562 cells by the ENCODE consortium [28]. Since Sp1 and Sp2 binding sites largely overlap, it was concluded that Sp1 and Sp2 binding motifs (i.e. GC boxes) are indistinguishable [29]. We believe that this interpretation of the ChIP-seq data results from the structural similarity of Sp1 and Sp2, and from Sp2´s capacity to bind GC rich oligonucleotides in vitro [18,19]. Admittedly, we also fell into this trap when we initially analyzed our Sp2 ChIP-seq data [18]. These misinterpretations emphasize the limitations of genomics data integration and highlight the importance of performing additional detailed experimental analysis.
The data presented here revealed fundamental different modes of binding site selection of Sp1/Sp3 and of Sp2, and also allow different interpretations of the Sp1-Nf-y and Sp2-Nf-y interactions. Sp1 and Sp3 ChIP-seq peaks locate to GC boxes that are often accompanied by Nf-y binding sites (see Fig. 2) strongly suggesting that Sp1/Sp3 and Nf-y co-bind to neighboring sites in the genome. This is consistent with earlier reports showing synergistic activation by Sp1 and Nf-y and a direct interaction between Sp1 (and Sp3) and Nf-ya [30,31,32]. In contrast, the majority of Sp2 locates at CCAAT boxes and binding is independent of the presence of a nearby GC box, and, most importantly, independent of the zinc finger domain.
The localization of Sp2 at CCAAT motifs led us to detail the interaction network between Sp2 and Nf-y on a genome-wide scale. The arrangement of CCAAT motifs in regions that are bound by both factors differ from sites that are bound by Nf-y but not by Sp2. Sp2-Nf-y interactions occur predominantly at tandem CCAAT boxes, whereas single CCCAT boxes are mostly bound by Nf-y but not by Sp2. Utilizing Sp2ko cells lacking any Sp2 DNA-binding activity, and an RNAi approach to interfere with Nf-y binding, we have explored the interaction between Nf-y and Sp2 recruitment in an unbiased manner. Genomic binding analysis revealed a widespread attenuation of Nf-y loading in cells lacking Sp2. Vice versa, reduced binding of Nf-y led to attenuation of Sp2 binding. Importantly, the reduction of Sp2 binding is specific to elements that are also bound by Nf-y, thereby providing support for a direct effect of Nf-y at shared sites. Given that multiple CCAAT boxes in a promoter region are simultaneously occupied by Nf-y, the particular placement of Nf-y complexes might be essential for direct or indirect interactions with Sp2. In line with this, we observed a general agreement between the strength of the Sp2 ChIP-seq signals and the number of CCAAT motifs. Particularly, the strongest Sp2 peaks contain several pairs of CCAAT motifs.
In a recent publication it was shown that Nf-y promotes binding of pluripotency transcription factors such as Oct4 or Sox2 at enhancers in murine ES cells by facilitating a permissive chromatin conformation [33]. Nf-y´s role in promoting the binding of Sp2 could be by a similar mechanism. However, unlike the pluripotency factors, Sp2 also potentiates binding of Nf-y at sites of co-localization. Whether Sp2 facilitates recruitment of Nf-y or whether it stabilizes binding of Nf-y at shared sites remains elusive at this stage.
The mode of interaction between different transcription factors has been classically categorized into DNA-binding dependent (co-binding) and DNA-binding independent, whereby one transcription factor binds to another that, in turn, binds to DNA (tethering). Tethering to chromatin has been reported for several transcription factors including recruitment of the glucocorticoid receptor by AP1 [34,35] or STAT3 [36] and the estrogen receptor alpha by Runx1 [37]. Likely, tethering mechanisms are also involved in the recruitment of SCL/TAL1 [38] and KLF3 [39] to a subset of their binding sites in vivo. The sequence adjacent to CCAAT boxes at which Sp2 is located is highly variable and does not contain a particular sequence motif. Consistently, the N-terminal region of Sp2 does not contain an obvious structural motif that might interact with a specific DNA sequence. It can be proposed that Nf-y binds to the CCAAT box through base-specific contacts. Sp2 would then be recruited by interaction with Nf-y or with additional factors. However, this simple tethering model does not explain the attenuated binding of Nf-y in Sp2ko cells at shared sites (Fig. 9), and the exceptionally large Sp2 ChIP-seq peaks and high ChIP-qPCR values (more than 10% of input on highly occupied promoters; see Figs. 3, 5, 9) particularly as formaldehyde preferentially crosslinks proteins to DNA. Therefore, we envisage an alternative mechanism. Potentially, promoters bound by Nf-y, particularly those bound by two or more Nf-y complexes, adopt a particular conformation that allows the basic N-terminal region of Sp2 to interact, directly or indirectly, with two Nf-y complexes simultaneously, and additionally with the DNA backbone in a sequence-independent manner (Fig. 10). Such a scenario would explain the mutual dependency of Sp2 and Nf-y binding to common sites.

Finally, a small fraction of the Sp2 binding sites does not contain CCAAT boxes and are not co-occupied by Nf-y. Nevertheless, binding of Sp2 to these sites is also mediated by the N-terminal region suggesting that binding of Sp2 to these sites is by a similar mechanism. De novo motif discovery at these sites revealed enrichment of the Ets transcription factor-binding motif GGAAG. Future work aims to identify the Ets factor that occupies these sites. A promising candidate is the widely expressed heterodimeric Ets transcription factor Gabp [40] that binds as a GABPalpha2beta2 heterotetramer complex to DNA containing two tandem GGAAG sites [41].
In conclusion, the data provided in this report challenge the prevailing view that the transcription factors Sp1/Sp3 and Sp2 regulate transcription by binding to similar promoter elements via their zinc finger domains. Instead, Sp1/Sp3 and Sp2 have distinctive binding landscapes, and their modes of genomic binding site selection are completely different. Collectively, our findings uncover strikingly different recruitment mechanisms of very similar transcription factors, and add another crucial level of detail to the current model of transcription factor binding to chromatin.
Materials and Methods
Antibodies
For Western blotting and ChIP of Sp1, Sp2 and Sp3 in MEFs, we used homemade rabbit antibodies [7,42] affinity-purified against the respective recombinant Sp factor. Anti-Sp3 antibodies (Santa Cruz, sc-644) were used for the Western blot shown in Fig. 3B, and anti-Sp2 antibodies (Santa Cruz, sc-643) for ChIP-seq of Sp2 in HEK293 cells. Additional antibodies: Anti-Nf-ya (Santa Cruz, sc-10779), anti-Nf-yb (Genespin, PAb001), anti-Nf-yc (Santa Cruz, sc-7715-R), anti-Flag M2 (Sigma, F3165), anti-Tubulin (Millipore, MAB3408).
Construction of retroviral vectors and retroviral transduction
Retroviral expression plasmids for 3xFlag-Sp2 and 3xFlag-Sp3 mutants were generated by restriction cloning of PCR fragments into a pBABE3xFlag-puro plasmid. Primer sequences used for PCR can be found in S1 Table. The production of virus stocks, infection of MEFs and the selection of transduced Sp2ko and Sp3ko MEFs were as described [7].
Cell growth conditions and generation of cell lines
MEFs were cultured in a 1 : 1 mixture of Dulbecco’s modified Eagle’s medium—high Glucose (PAA) and HAM’s F-10 (PAA) supplemented with 10% (v/v) fetal calf serum (PAA) and 1% Penicillin-Streptomycin. Wild type and Sp3ko MEFs, and MEFs with floxed Sp2 alleles (Sp2fl/fl) were isolated from E13.5 embryos using standard methods and subsequently immortalized by serial passages. Sp2ko MEFs were obtained by retroviral transduction of pBABE-Cre-neo as described [7]. Sp2ko MEFs have severely impaired proliferation rates [7]. Cells that escaped growth inhibition over time were used for rescue experiments.
Immunohistochemistry
Immunofluorescence and microscopy was performed as described [43]. In brief, 5x104 MEF cells expressing 3xFlag-Sp2 mutants were grown on coverslips in 6-well plates overnight. Cells were fixed in 4% PFA/PBS for 25 min, permeabilized in 0.2% TritonX-100/PBS for 20 min, and blocked with 3% BSA/PBS for 1 hr. Incubation of the anti-Flag M2 antibody (Sigma, F3165, 1 : 800 dilution) was for 1 hr at RT. Secondary antibody incubation (anti-mouse AlexaFluor568, Invitrogen, A10037, 1 : 500 dilution) was performed for 1 hr at RT in the dark. After a final washing step, coverslips were mounted onto glass slides using Vectashield mounting medium with DAPI (Vector Laboratories, Inc.).
Knockdown of Nf-y subunits
For RNAi-mediated depletion of mouse Nf-ya, Nf-yb and Nf-yc, pools of four On-target plus siRNAs (GE Dharmacon) were used (LU-065522, LU-046072, LU-060374). The siGenome non-targeting siRNA #1 (D-001210–01) was used as unspecific siRNA control. Wild type MEFs on 15 cm plates were transfected with 24 nM siRNA using Oligofectamine (Invitrogen). Three days post-transfection 2x106 cells were replated, and transfected a second time. Additional three days later, cells were collected and cross-linked chromatin was prepared. To monitor knockdown efficiency at the protein level, a chromatin sample was incubated with an equal amount of 2xLaemmli buffer at 100°C for one hour and subsequently analyzed by western blotting. To monitor knockdown efficiency at the transcript level, RNA was isolated using the RNeasy Mini kit (Qiagen) and expression of Nf-ya, Nf-yb and Nf-yc was analyzed by RT-qPCR.
RT-qPCR
Expression analysis by quantitative RT-qPCR was performed as described in [44] using gene-specific primers (S1 Table).
ChIP-qPCR
ChIP experiments were performed as described [18] using the One Day ChIP kit (Diagenode). For a sequential ChIP of Sp2 and Nf-yb, the precipitated material of a standard ChIP was eluted twice from the beads with 100 mM NaHCO3, 1% SDS, 10 mM DTT for 30 min at 37°C. Eluates were diluted 1 : 50 with ChIP buffer and subsequently subjected to a second ChIP in accordance with the One Day ChIP kit manual performing an overnight antibody incubation at 4°C. Enrichment was calculated relative to the input of the first ChIP. Primer sequences for ChIP-qPCRs are listed in S1 Table.
ChIP-seq and data analysis
For ChIP-seq experiments, four to six individual ChIPs were pooled, and precipitated DNA was purified on QIAquick columns (Qiagen). Five nanograms of DNA were used for indexed next generation sequencing library preparation using the MicroPlex library preparation kit (Diagenode) in accordance with the manufacturer’s instructions. Library purification was performed with AMPure magnetic beads (Beckman Coulter) as described in the MicroPlex kit manual. Libraries were quantified on a Bioanalyzer (Agilent Technologies) and subsequently sequenced on an Illumina HiSeq1500 platform, rapid-run mode, single-read 50 bp (TruSeq Rapid SR Cluster Kit—HS, TruSeq Rapid SBS Kit—HS—50 cycle) according to manufacturer´s instructions. An overview of the various ChIP-seq results is shown in S2 Table.
Raw ChIP-seq data were aligned to the mouse genome assembly mm10 or the human genome assembly hg19 using Subread 1.3.3-p3 [45]. Reads were filtered to have at most 5 mismatches to the reference, indels up to 5 bp and to occur at exactly one position in the genome. Peak calling was performed using MACS 1.4 [46] with default parameters. Raw and normalized (to 1 million uniquely aligned reads) read counts were annotated, and peaks were filtered to have at least 30 raw reads and a signal to background (IgG, Sp2ko or Sp3ko) normalized read ratio of at least 3. Further analysis of peaks such as association with transcripts in the vicinity, classification of genomic position and Venn diagram generation was performed using a custom Python based pipeline. Genome annotation data from Ensembl revision 74 [47] was used. Position and characteristics of ChIP-seq peaks are listed in S3 Table. Venn diagrams were calculated by building the union of the datasets involved, and assigning each union-peak to a region by requiring at least a one-basepair overlap with the input regions. De novo motif search including central motif analysis [21] was performed with MEME-ChIP version 4.9.1 [48] using sequences surrounding peak summits (+/ - 150 bp). Elongating reads by 200 bp, and determining the position of highest overlap defined summits.
Data deposition
Raw sequencing data were deposited at ArrayExpress under accession number E-MTAB-2970.
Supporting Information
Zdroje
1. Suske G, Bruford E, Philipsen S (2005) Mammalian SP/KLF transcription factors: bring in the family. Genomics 85 : 551–556. 15820306
2. Philipsen S, Suske G (1999) A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res 27 : 2991–3000. 10454592
3. Lomberk G, Urrutia R (2005) The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem J 392 : 1–11. 16266294
4. Kaczynski J, Cook T, Urrutia R (2003) Sp1 - and Kruppel-like transcription factors. Genome Biol 4 : 206. 12620113
5. Schaeper ND, Prpic NM, Wimmer EA (2010) A clustered set of three Sp-family genes is ancestral in the Metazoa: evidence from sequence analysis, protein domain structure, developmental expression patterns and chromosomal location. BMC Evol Biol 10 : 88. doi: 10.1186/1471-2148-10-88 20353601
6. Suske G (1999) The Sp-family of transcription factors. Gene 238 : 291–300. 10570957
7. Baur F, Nau K, Sadic D, Allweiss L, Elsasser HP, et al. (2010) Specificity protein 2 (Sp2) is essential for mouse development and autonomous proliferation of mouse embryonic fibroblasts. PLoS One 5: e9587. doi: 10.1371/journal.pone.0009587 20221402
8. Bouwman P, Göllner H, Elsässer HP, Eckhoff G, Karis A, et al. (2000) Transcription factor Sp3 is essential for post-natal survival and late tooth development. EMBO J 19 : 655–661. 10675334
9. Marin M, Karis A, Visser P, Grosveld F, Philipsen S (1997) Transcription factor Sp1 is essential for early development but dispensable for cell growth and differentiation. Cell 89 : 619–628. 9160753
10. Liang H, Xiao G, Yin H, Hippenmeyer S, Horowitz JM, et al. (2013) Neural development is dependent on the function of specificity protein 2 in cell cycle progression. Development 140 : 552–561. doi: 10.1242/dev.085621 23293287
11. Kim TH, Chiera SL, Linder KE, Trempus CS, Smart RC, et al. (2010) Overexpression of Transcription Factor Sp2 Inhibits Epidermal Differentiation and Increases Susceptibility to Wound - and Carcinogen-Induced Tumorigenesis. Cancer Res 70 : 8507–8516. doi: 10.1158/0008-5472.CAN-10-1213 20959487
12. Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, et al. (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464 : 721–727. doi: 10.1038/nature08869 20360735
13. Göllner H, Dani C, Phillips B, Philipsen S, Suske G (2001) Impaired ossification in mice lacking the transcription factor Sp3. Mech Dev 106 : 77–83. 11472836
14. Van Loo PF, Bouwman P, Ling KW, Middendorp S, Suske G, et al. (2003) Impaired hematopoiesis in mice lacking the transcription factor Sp3. Blood 102 : 858–866. 12676787
15. Van Loo PF, Mahtab EA, Wisse LJ, Hou J, Grosveld F, et al. (2007) Transcription factor Sp3 knockout mice display serious cardiac malformations. Mol Cell Biol 27 : 8571–8582. 17923686
16. Kingsley C, Winoto A (1992) Cloning of GT box-binding proteins: a novel Sp1 multigene family regulating T-cell receptor gene expression. Mol Cell Biol 12 : 4251–4261. 1341900
17. Moorefield KS, Yin H, Nichols TD, Cathcart C, Simmons SO, et al. (2006) Sp2 localizes to subnuclear foci associated with the nuclear matrix. Mol Biol Cell 17 : 1711–1722. 16467376
18. Terrados G, Finkernagel F, Stielow B, Sadic D, Neubert J, et al. (2012) Genome-wide localization and expression profiling establish Sp2 as a sequence-specific transcription factor regulating vitally important genes. Nucleic Acids Res 40 : 7844–7857. doi: 10.1093/nar/gks544 22684502
19. Moorefield KS, Fry SJ, Horowitz JM (2004) Sp2 DNA binding activity and trans-activation are negatively regulated in mammalian cells. J Biol Chem 279 : 13911–13924. 14726517
20. Krüger I, Vollmer M, Simmons DG, Elsässer HP, Philipsen S, et al. (2007) Sp1/Sp3 compound heterozygous mice are not viable: impaired erythropoiesis and severe placental defects. Dev Dyn 236 : 2235–2244. 17584888
21. Bailey TL, Machanick P (2012) Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40: e128. 22610855
22. Dolfini D, Zambelli F, Pavesi G, Mantovani R (2009) A perspective of promoter architecture from the CCAAT box. Cell Cycle 8 : 4127–4137. 19946211
23. Stielow B, Sapetschnig A, Krüger I, Kunert N, Brehm A, et al. (2008) Identification of SUMO-dependent chromatin-associated transcriptional repression components by a genome-wide RNA interference screen. Mol Cell 29 : 742–754. doi: 10.1016/j.molcel.2007.12.032 18374648
24. Stielow B, Sapetschnig A, Wink C, Krüger I, Suske G (2008) SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep 9 : 899–906. doi: 10.1038/embor.2008.127 18617891
25. Stielow B, Krüger I, Diezko R, Finkernagel F, Gillemans N, et al. (2010) Epigenetic silencing of spermatocyte-specific and neuronal genes by SUMO modification of the transcription factor Sp3. PLoS Genet 6: e1001203. doi: 10.1371/journal.pgen.1001203 21085687
26. Dolfini D, Gatta R, Mantovani R (2012) NF-Y and the transcriptional activation of CCAAT promoters. Crit Rev Biochem Mol Biol 47 : 29–49. doi: 10.3109/10409238.2011.628970 22050321
27. Nardini M, Gnesutta N, Donati G, Gatta R, Forni C, et al. (2013) Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell 152 : 132–143. doi: 10.1016/j.cell.2012.11.047 23332751
28. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, et al. (2012) The accessible chromatin landscape of the human genome. Nature 489 : 75–82. doi: 10.1038/nature11232 22955617
29. Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, et al. (2012) Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22 : 1798–1812. doi: 10.1101/gr.139105.112 22955990
30. Yamada K, Tanaka T, Miyamoto K, Noguchi T (2000) Sp family members and nuclear factor-Y cooperatively stimulate transcription from the rat pyruvate kinase M gene distal promoter region via their direct interactions. J Biol Chem 275 : 18129–18137. 10748033
31. Roder K, Wolf SS, Larkin KJ, Schweizer M (1999) Interaction between the two ubiquitously expressed transcription factors NF-Y and Sp1. Gene 234 : 61–69. 10393239
32. Lim K, Chang HI (2009) O-GlcNAcylation of Sp1 interrupts Sp1 interaction with NF-Y. Biochem Biophys Res Commun 382 : 593–597. doi: 10.1016/j.bbrc.2009.03.075 19302979
33. Oldfield AJ, Yang P, Conway AE, Cinghu S, Freudenberg JM, et al. (2014) Histone-Fold Domain Protein NF-Y Promotes Chromatin Accessibility for Cell Type-Specific Master Transcription Factors. Mol Cell 55 : 708–722. doi: 10.1016/j.molcel.2014.07.005 25132174
34. Schüle R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, et al. (1990) Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 62 : 1217–1226. 2169353
35. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, et al. (2011) Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43 : 145–155. doi: 10.1016/j.molcel.2011.06.016 21726817
36. Langlais D, Couture C, Balsalobre A, Drouin J (2012) The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell 47 : 38–49. doi: 10.1016/j.molcel.2012.04.021 22633955
37. Stender JD, Kim K, Charn TH, Komm B, Chang KC, et al. (2010) Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol 30 : 3943–3955. doi: 10.1128/MCB.00118-10 20547749
38. Kassouf MT, Hughes JR, Taylor S, McGowan SJ, Soneji S, et al. (2010) Genome-wide identification of TAL1's functional targets: insights into its mechanisms of action in primary erythroid cells. Genome Res 20 : 1064–1083. doi: 10.1101/gr.104935.110 20566737
39. Burdach J, Funnell AP, Mak KS, Artuz CM, Wienert B, et al. (2014) Regions outside the DNA-binding domain are critical for proper in vivo specificity of an archetypal zinc finger transcription factor. Nucleic Acids Res 42 : 276–289. doi: 10.1093/nar/gkt895 24106088
40. Rosmarin AG, Resendes KK, Yang Z, McMillan JN, Fleming SL (2004) GA-binding protein transcription factor: a review of GABP as an integrator of intracellular signaling and protein-protein interactions. Blood Cells Mol Dis 32 : 143–154. 14757430
41. Chinenov Y, Henzl M, Martin ME (2000) The alpha and beta subunits of the GA-binding protein form a stable heterodimer in solution. Revised model of heterotetrameric complex assembly. J Biol Chem 275 : 7749–7756. 10713087
42. Hagen G, Müller S, Beato M, Suske G (1994) Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J 13 : 3843–3851. 8070411
43. Sapetschnig A, Koch F, Rischitor G, Mennenga T, Suske G (2004) Complexity of translationally controlled transcription factor Sp3 isoform expression. J Biol Chem 279 : 42095–42105. 15247228
44. Stielow C, Stielow B, Finkernagel F, Scharfe M, Jarek M, et al. (2014) SUMOylation of the polycomb group protein L3MBTL2 facilitates repression of its target genes. Nucleic Acids Res 42 : 3044–3058. doi: 10.1093/nar/gkt1317 24369422
45. Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41: e108. doi: 10.1093/nar/gkt214 23558742
46. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. doi: 10.1186/gb-2008-9-9-r137 18798982
47. Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, et al. (2013) Ensembl 2013. Nucleic Acids Res 41: D48–55. doi: 10.1093/nar/gks1236 23203987
48. Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27 : 1696–1697. doi: 10.1093/bioinformatics/btr189 21486936
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2015 Číslo 3
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs