
Identification of Anti-virulence Compounds That Disrupt Quorum-Sensing Regulated Acute and Persistent Pathogenicity
Antibiotic resistant and tolerant bacterial pathogens are responsible for acute, chronic and persistent human infections recalcitrant to any current treatments. Therefore, there is an urgent need to identify new antimicrobial drugs that will help circumvent the current antibiotic resistance crisis. Bacterial pathogens often develop resistance to antibiotic drugs that target bacterial growth or viability. In contrast, strategies that specifically target virulence pathways non-essential for growth could limit selective resistance, and thus are candidates for the development of next-generation antimicrobial therapeutics. In this study we target the bacterial communication system MvfR (PqsR), which is known to control virulence of the opportunistic bacterial pathogen Pseudomonas aeruginosa. We identified and improved upon new small molecules that effectively silence the MvfR communication system, and as a result block P. aeruginosa virulence both in vitro and in vivo. Moreover, these new compounds are the first known to restrict the ability of bacteria to form antibiotic-tolerant cells and consequently proved to be very effective at preventing persistent infection in a mammalian infection model. Because of their ability to simultaneously block acute and persistent infections, these new molecules may provide a very strong basis for the development of next generation antimicrobials.
Published in the journal:
Identification of Anti-virulence Compounds That Disrupt Quorum-Sensing Regulated Acute and Persistent Pathogenicity. PLoS Pathog 10(8): e32767. doi:10.1371/journal.ppat.1004321
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004321
Summary
Antibiotic resistant and tolerant bacterial pathogens are responsible for acute, chronic and persistent human infections recalcitrant to any current treatments. Therefore, there is an urgent need to identify new antimicrobial drugs that will help circumvent the current antibiotic resistance crisis. Bacterial pathogens often develop resistance to antibiotic drugs that target bacterial growth or viability. In contrast, strategies that specifically target virulence pathways non-essential for growth could limit selective resistance, and thus are candidates for the development of next-generation antimicrobial therapeutics. In this study we target the bacterial communication system MvfR (PqsR), which is known to control virulence of the opportunistic bacterial pathogen Pseudomonas aeruginosa. We identified and improved upon new small molecules that effectively silence the MvfR communication system, and as a result block P. aeruginosa virulence both in vitro and in vivo. Moreover, these new compounds are the first known to restrict the ability of bacteria to form antibiotic-tolerant cells and consequently proved to be very effective at preventing persistent infection in a mammalian infection model. Because of their ability to simultaneously block acute and persistent infections, these new molecules may provide a very strong basis for the development of next generation antimicrobials.
Introduction
Antibiotic-resistant and tolerant microbes mediate acute, persistent, chronic, and/or relapsing human infections [1]–[3]. Such infections occur worldwide, affect all sectors, cause physical and emotional suffering, impose high financial costs on patients and healthcare systems, and are refractory to current anti-infective drugs. As such, identification of new molecular targets and corresponding compounds to restrict multidrug-resistant (MDR) and antibiotic-tolerant (AT) infections will substantively benefit human healthcare.
Bacterial pathogens often develop resistance to antibiotic drugs that target bacterial growth or viability. In contrast, strategies that specifically target virulence pathways that are non-essential for growth could limit selective resistance, and thus are candidates for the development of next-generation antimicrobial therapeutics. One candidate pathway is quorum sensing (QS), a cell-to-cell density-dependent communication system mediated via the production of and regulation by low molecular weight signaling molecules. QS, which is evolutionarily conserved throughout eubacteria and archaebacteria, is crucial for the development and maintenance of acute and chronic/persistent human infections as well as the commonly observed antibiotic tolerance of many pathogenic bacteria [4]–[10]. As such, anti-virulence compounds that specifically target QS could have a major impact on the control and treatment of a wide-range of acute and persistent bacterial infections [11]–[13].
P. aeruginosa is a wide-spread opportunistic human pathogen responsible for acute and chronic/persistent infections that readily develop multi-drug resistance to clinical antibiotics, and often evade clinical treatment [1]–[3]. P. aeruginosa has three distinct QS systems mediated by cell-to-cell signals including the acyl-homoserine lactones (HSL) 3-oxo-C12-HSL and C4-HSL, respectively produced by the las and rhl QS systems; and the 4-hydroxy-2-alkylquinolines (HAQs), produced by the mvfR (pqsR) QS system [14]. MvfR is a LysR-type transcriptional regulator (LTTR) that directs the synthesis of ∼60 low molecular weight HAQ molecules, including its positive regulatory ligands 4-hydroxy-2-heptylquinoline (HHQ) and 3,4-dihydroxy-2-heptylquinoline (PQS); and the non-HAQ, 2-aminoacetophenone (2-AA) [7], [15]–[16]. LTTRs control the expression of a diverse array of virulence regulons in Gram-negative and Gram-positive pathogens, and are the largest family of homologous regulators in prokaryotes [17].
While all three P. aeruginosa QS systems are required for full pathogenicity in mammalian hosts [18]–[21], the lasR pathway is often inactivated in isolates from cystic fibrosis (CF) patients, and thus it may be nonessential for chronic/persistent infections. This inactivation is due to mutations in LasR itself [22], [23], and may be due to specific MvfR-regulated functions [7]. Conversely, MvfR is essential for full virulence in several host models [19], [24], [25], and clinical isolates with mvfR mutations have not been identified. MvfR binds to and activates the pqs operon, which encodes enzymes for the synthesis of HAQs, including PQS and HHQ [15], [16], [26]; and for MvfR-regulated small molecules, including 2-AA. These molecules are produced in human tissues and function in pathogenicity [27], [28]. Both HHQ and PQS bind to and activate MvfR [16], [26] to lead to the production of MvfR-regulated virulence factors that promote acute infections [25], [29]–[31]. 2-AA, which is produced in human tissues [32], signals changes in both bacterial [7] and host pathways [6], [33]. Some of the affected pathways underlie the development and maintenance of chronic/persistent infections, including functions that promote antibiotic tolerance [8], long-term survival and persistence [7], and modulation of host functions that promote pathogen tolerance [6].
Antibiotic-tolerant (AT) cells underlie bacterial persistence and correspond to sub-populations that survive lethal concentrations of antibiotics. AT cells are implicated in the clinical failure of antibiotic therapy, and may populate and/or be responsible for persistent infections that can be the source of latent, chronic, or relapsing infections that are suppressed but not eradicated by antibiotics [34]–[36].
MvfR, due to its central role in both acute and chronic/persistent infections, is a potential target for the development of new anti-microbial drugs, especially as it is nonessential for cell viability or growth. Here we identify robust quorum sensing inhibitors (QSI) that inhibit the MvfR virulence regulon via binding to the MvfR regulatory protein; are highly efficacious in disrupting MvfR-dependent cell-to-cell communication in vivo; and limit P. aeruginosa infections and lethality in mice. Moreover, these are the first identified compounds that restrict the formation of antibiotic-tolerant persister cells, and consequently, that restrict P. aeruginosa persistent infections in mice. These molecules, which belong to a chemical family previously unrecognized for MvfR inhibitory activity, provide for the development of effective clinical therapeutics to limit and eradicate acute and chronic/persistent multi-drug resistant infections.
Results
High-throughput whole-cell screening identifies novel potent MvfR-regulon inhibitors with a benzamide-benzimidazole chemical backbone
We used a whole cell high-throughput screen (HTS) to identify compounds that inhibit MvfR regulon activity without perturbing cell viability or growth (Fig. 1, S1 and S2b). We screened a chemical library of 284,256 low molecular weight compounds for inhibition of pqsA expression using a reporter consisting of the pqsA promoter fused to the sacB gene (Fig. S1) [7], [37]. In this screen, a solvent control or non-inhibitory compound results in bacterial death when sucrose is present in the culture media due to its conversion to toxic levans by the sacB gene product [37], while compounds that inhibit pqsA promoter expression, and thus HAQ synthesis, permit bacterial growth.

The MvfR-regulon inhibitory compounds initially identified belong to 7 distinct chemical families (Fig. 1). The most effective inhibitors were verified via a second screen for pqsA promoter repression using a pqsA-GFP [38] reporter construct, which yielded 39 candidate compounds (Figs. S1 and S2a). These inhibitors were further analyzed by functional assays for reduced HAQ production, including HQNO, and the MvfR ligands HHQ and PQS; and for reduced levels of pyocyanin, an MvfR-regulated virulence effector. Figure 1 presents the structures and LC/MS results for 17 compounds that completely eliminated pqsA-GFP mediated fluorescence, greatly reduced HHQ, PQS, and HQNO levels at 50 µg/mL, and notably, did not impact bacterial growth (Fig. S2). Some of these compounds also eliminated or greatly reduced pyocyanin levels (Fig. 1). Strikingly, 8 of these compounds (labeled in red) share a benzamide-benzimidazole (BB) backbone, consisting of a substituted benzamide moiety and endocyclic aromatic amines. These BB inhibitors also increased anthranillic acid (AA) in culture (data not shown), likely via its non-utilization and subsequent accumulation [15], [39]. The other 9 inhibitors presented in Figure 1 are unrelated to the BB inhibitors and do not share any common structure or distinct feature between them. Also, 12 of the total 17 inhibitors reduced pyocyanin to less than 50%, while the BB compound M4 and the non-BB compounds M21, M29, M31, and M32, did not (Fig. 1). All BB inhibitors identified from the HTS, except for M24, reduced HHQ and PQS levels to ≤15%, and M4, M23, M26, M27, and M34 were effective at ≤10 µM (Fig. 2). This concentration is 150 fold lower than the effective concentrations for previously identified MvfR-regulon inhibitors [40].

Structure-activity relationship analysis identifies improved 2nd generation benzamide-benzamidazole MvfR-regulon inhibitors
We performed a structure-activity relationship (SAR) analysis to define 2nd generation BB derivatives with enhanced MvfR-regulon inhibitory activity, as determined by repression of HAQ and pyocyanin production at 10 µM and 1 µM (Fig. 2). Starting with M56, increased inhibitor activity was obtained by adding an electron withdrawing group, such as a nitro, to position 5 of the benzimidazole moiety, to give M52; or an electron-releasing group to the benzamide para position, to give M55. As such, we focused on compounds with a nitro substituted benzimidazole ring and a para substituted benzamide ring. Derivatives having a methoxy (M61), trifluoromethoxy (M58), chloro (M51), or a cyano (M62) para group showed increased inhibitory activity at ≤1 µM IC50 (Fig. 2, white). For further optimization, we synthesized BB derivatives containing a nitro substituted benzimidazole ring and a para-bromo (M50), iodo (M59), or phenoxy (M64) substituted benzamide ring (Fig. 2, green). Also, the BB thioether bond was critical for inhibition in M59, as replacing it with a methylene eliminates activity (data not shown). M64 was the most effective 2nd generation inhibitor for reducing PQS, HHQ, and pyocyanin production, with respective IC50 of 200 nM, 350 nM, and 300 nM (Fig. S3).
MvfR regulon inhibitors restrict the formation of P. aeruginosa antibiotic tolerant cells
2-AA, an abundant MvfR-regulated non-HAQ small molecule, promotes P. aeruginosa antibiotic-tolerant (AT) cell formation [8], [41] and bacterial persistence in infected flies [7] and mice [6]. Figure 3 shows that the MvfR BB inhibitors prevented both 2-AA synthesis and AT cell accumulation, suggesting their potential to limit P. aeruginosa chronic/persistent infections.

Figure 3a shows that the 6 most potent BB inhibitors of HAQ and pyocyanin production (M34, M50, M51, M59, M62 and M64) dramatically reduced 2-AA production; while two potent HAQ non-BB inhibitors, M29 and M31, conversely, slightly increased 2-AA production (Fig. 3a). Figure 3b shows that all of the BB compounds that decreased 2-AA production also decreased the number of persister cells tolerant to the β-lactam antibiotic meropenem while alternatively, M29 increased persisters, perhaps via increased 2-AA.
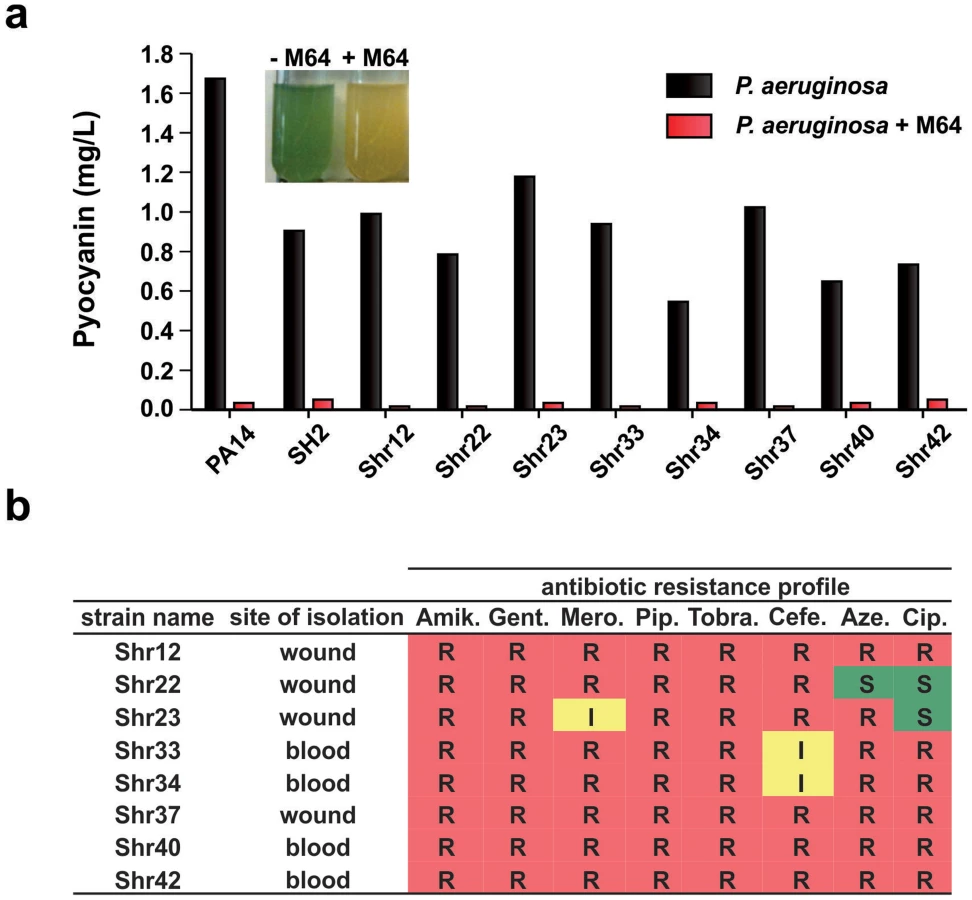
We focused on the BB compounds that restrict persister formation. Figure 3c shows the generalized anti-persister efficacy of M64, as it limits formation of antibiotic-tolerant persisters to other antibiotic classes, including quinolones (ciprofloxacin, and levofloxacin), and aminoglycosides (amikacin). In addition, M64 inhibited pyocyanin production in several P. aeruginosa clinical isolates (Fig. 4a), including multidrug or pan-resistant strains (Fig. 4b), suggesting its potential for the development of anti-infective reagents against recalcitrant MDR strains.
Mechanism of MvfR regulon inhibition
The mechanism of action of the most potent MvfR inhibitors is not obvious, as their common BB backbone is unrelated to MvfR ligands or the biosynthetic precursors or intermediates of these ligands. As these compounds decrease HAQ and 2-AA production, they might target the MvfR regulatory protein, or alternatively, the pqs operon enzymes that mediate HAQ and 2-AA biosynthesis [15]. To this end, we asked if the MvfR-regulon inhibitors reduce HAQs, as assessed by LC/MS, in an isogenic mvfR mutant strain that constitutively expresses the pqsABCD genes, and thus has MvfR-independent HAQ production. Figure 5a shows that the most potent BB inhibitors (M51, M34, M62, M50 and M64) did not alter HAQ levels compared to the solvent control, suggesting that they target MvfR or another upstream regulatory component. In contrast, the non-BB inhibitor, M29, reduced HHQ, PQS, and HQNO levels, and increased DHQ (Fig. 5a). Since PqsA and PqsD are necessary and sufficient for DHQ production [42], [43], M29 may inhibit the PqsB and/or PqsC enzymes that are required for HHQ and PQS, but not DHQ or 2-AA production [7].

An essential step of pqsA promoter activation is the binding of the MvfR protein to specific DNA residues within the pqs promoter, which is enhanced by PQS or HHQ [16]. To determine whether M64 disrupts this binding, PA14 cells expressing MvfR fused to a vesicular stomatitis virus glycoprotein (VSV-G) epitope at the C-terminus were grown with and without M64, and the MvfR–DNA complex was isolated via chromatin immunoprecipitation (ChIP). The co-precipitated DNA was quantified by qPCR [19], [44]. Figure 5b shows that M64 decreased MvfR binding to the pqsA promoter by ∼10-fold and blocked the PQS – mediated increase in MvfR binding (Fig. 5b). As shown in Figure S5, PQS and/or M64 addition did not affect MvfR levels.
Figure 5 demonstrates that the M64 molecular target is MvfR itself, rather than an upstream component. Isothermal titration calorimetry (ITC) showed that this binding has a ∼1∶1 stoichiometry for M64 and the MvfR co-inducer binding domain, with a KD = 5.4 nM (Fig. 5c). This binding likely prevents MvfR binding to the pqsA promoter to inhibit MvfR regulon activation.
MvfR regulon inhibitors attenuate P. aeruginosa acute virulence
MvfR QS is a target pathway for the development of new anti-infective reagents, as it controls a large regulon of virulence functions [16], [25] and is required for pathogenicity in evolutionary distinct hosts [19], [20], [24], [31], [45]–[47]. To this end, we asked if our MvfR inhibitors limit bacterial virulence in murine macrophages (Fig. 6) and in mice (Figs. 7–9). Figure 6 shows that the BB inhibitors significantly reduced PA14 cytotoxicity in Raw 264.7 macrophages. M64 was the most effective compound and was not cytotoxic to the macrophages. 76% of PA14-infected cells survived in the presence of M64 compared to only 36% survival in the absence of this compound (p<0.01). In addition, mvfR mutant cells were less cytotoxic than wild-type PA14, and M64 did not rescue this cytoxicity, further confirming that it targets the MvfR pathway.

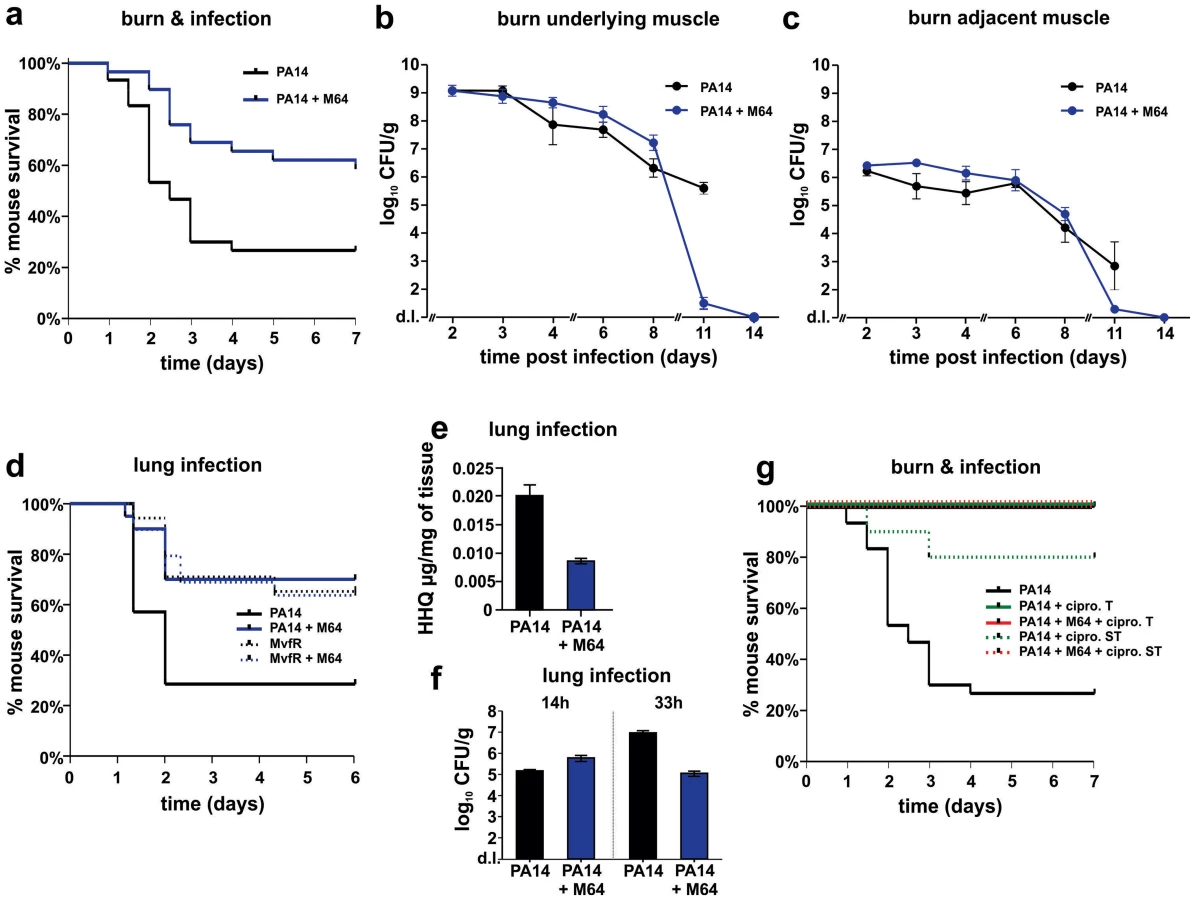


Two distinct in vivo murine models of acute infection (thermal injury and lung infection) were used to ask if M64 is therapeutically efficacious for P. aeruginosa-mouse pathogenesis [40], particularly for highly virulent strains exemplified by PA14. Burned and infected mice that were injected with M64 beginning 6 h post-infection followed by injections twice daily through day 6, exhibited increased survival compared to control mice (p<0.001), demonstrating M64's anti-infective efficacy in mice to reduce PA14 acute virulence (Fig. 7a). This increased host survival was not due to reduced bacterial loads, as both treated and untreated animals had similar CFUs at the infection site or at the adjacent muscle up to 6 days post-infection (Fig. 7b and 7c). As such, M64 directly reduced acute PA14 virulence in mice, likely by inhibiting virulence functions, as opposed to reducing bacterial loads. Using this model we compared the M64 sensitivity of colonies isolated from infected versus infected and M64 treated animals. M64 sensitivity was assessed at day 11 post-infection and treatment by measuring M64 IC50 for pyocyanin production in 10 remaining colonies per animal (n = 3 animals per group), reasoning that mutants resistant to M64 would still produce pyocyanin in the presence of M64. No difference in M64 IC50 was observed in any of the tested colonies, suggesting that resistance does not arise at least by day 10 post-treatment (data not shown).
The in vivo anti-infective activity of M64 was further confirmed in a murine acute lung infection model (Fig. 7d), as M64 significantly reduced the mortality of PA14 infected mice by ∼2.5-fold (p<0.05), comparable to that of animals infected with attenuated mvfR mutant bacteria. M64 did not further rescue the reduced mortality of mvfR-infected mice (Fig. 7d). In addition, in vivo HHQ levels were significantly decreased in infected animals treated with M64 (p<0.01) (Fig. 7e) and the pulmonary bacterial loads were unchanged in treated versus untreated animals at 14 h post-infection. These results suggest that the in vivo specificity of M64 to inhibit MvfR regulon function reduces virulence while not affecting bacterial viability. Nonetheless, Figure 7f shows that bacterial loads in the lungs were significantly reduced in the treated animals by 33 h post-infection, suggesting that M64 treatment facilitated host clearance. A similar effect is also observed in the muscle samples from burned and infected mice treated with M64 (Fig. 7b and 7c).
M64 combined with a sub-therapeutic (ST) dose of ciprofloxacin in the thermal injury and infection model resulted in 100% survival of PA14-infected mice versus 80% survival for mice receiving ST ciprofloxacin alone (p<0.001), suggesting an additive effect of M64 and ciprofloxacin (Fig. 7g). This effect is not due to altered ciprofloxacin minimal inhibitory concentration (MIC) (Table S1), and instead may be the consequence of differences in the mode of actions of these two compounds. Indeed, as the ciprofloxacin ST dose did not kill all of the PA14 cells, M64 might have reduced the virulence of the remaining cells to be cleared by the host immune system. Despite treatment ending at day 6, M64 monotherapy led to a dramatic decrease in bacterial CFUs in both muscle samples of burned and infected mice post-day 8, with complete bacterial tissue clearance by day 14 (Fig. 7b and 7c), demonstrating efficacy of M64 without the combinatorial antibiotic. None of the infected but untreated animals survived past day 11; hence no further samples could be taken from this population.
M64 attenuates macrophage accumulation at the infection site
We used a Magnetic Resonance Imaging (MRI) technique to evaluate M64 anti-infective efficacy in live animals by monitoring accumulation of macrophages, which are a marker of host anti-pathogen functions at active infection sites. We introduced a novel combination of off resonance imaging (ORI) positive-contrast MRI and T2ρ relaxation in the rotating frame (ORI-T2ρ) for positive-contrast MR imaging of ultra-small superparamagnetic iron-oxide (USPIO) nanoparticles. This technique exploits the chemical shift induced by USPIO nanoparticles engulfed by macrophages to nearby water molecules. Macrophages accumulate at the P. aeruginosa infection site in response to bacterial virulence factors and host immune response functions [48]. Figure 8 demonstrates, via in vivo MRI, that M64 reduced macrophage accumulation at the P. aeruginosa infection site in the murine burn and infection model, corroborating its anti-infective potential. M64 is not cytotoxic to murine macrophages in culture, supporting the notion that the reduced macrophage accumulation is via inhibition of PA14-mediated inflammation rather than macrophage killing.
M64 inhibits P. aeruginosa persistence in infected mice
Bacterial AT cells are likely a key component of latent, chronic, persistent, and relapsing infections, as they are a reservoir for re-initiation of active infections, and have been implicated in antibiotic treatment failures and host mortality [36], [49], [50]. To this end, new clinical therapies are needed that limit the ongoing presence of AT cells in infected hosts. Our inhibitors prevent 2-AA synthesis, which promotes the formation of P. aeruginosa persister cells [8], and persistence in vivo [6], [7].
We developed a mouse infection model to assess P. aeruginosa persistence in vivo following antibiotic treatment, and to evaluate M64 in vivo efficacy to reduce PA14 persistence in infected animal tissues, plus and minus combinatorial antibiotic therapy. Using this model, we demonstrate in Figure 9 that M64 inhibits persistence in the infected host, suggesting its broad anti-infective potential. No bacterial CFUs were detected in muscle tissue directly below or adjacent to the host infection site 4 days post-infection in animals treated with ciprofloxacin and M64, or with ciprofloxacin alone. Conversely, CFUs reappeared 6 days post-infection in the antibiotic-only treated animals, but not in those receiving antibiotic plus inhibitor. The co-injected animals exhibited minimal to no CFUs 6–8 days post-infection, with complete clearance by day 11 in the rectus abdominus muscle directly below or adjacent to the infection site, while the CFUs in the antibiotic-only animals were ∼103–104 6–8 days post-infection and ∼100 cells by day 14 (Fig. 9a and 9b). These CFUs were confirmed to be antibiotic sensitive, as cultures inoculated from single surviving colonies from animals at days 6, 8, and 11 had identical ciprofloxacin MICs and killing curves compared to the parental PA14 culture (data not shown).
These data demonstrate the additive effect of M64 on ciprofloxacin to fully clear infection and indicate that M64 prevents tissue re-colonization after antibiotic treatment is stopped. This effect is not due to altered ciprofloxacin MIC (Table S1), further substantiating the anti-persistence efficacy of the MvfR regulon inhibitors.
Discussion
The ideal anti-infective to treat bacterial infections in human patients should: 1) be highly specific to and inhibit a molecular pathway that is required for virulence in acute, chronic, and persistent infections; 2) be efficacious against divergent pathogens, by targeting an essential, evolutionarily conserved, and wide-spread virulence factor or pathway; 3) minimize selective resistance by not disrupting pathogen growth or viability; 4) inhibit the occurrence of AT pathogen cells; 5) be highly robust, and function at nM concentrations or less; 6) be non-toxic to and metabolically stable in host and pathogen cells; and 7) be affordably synthesized and administered. The novel compounds identified here exhibit several of these characteristics, and as such, provide a foundation for the development of next-generation anti-infective therapeutics.
Bacterial QS systems are candidate targets for the development of more effective drugs to treat acute and chronic or persistent bacterial infections, as QS pathways control the coordinated expression and production of a wide-array of virulence factors in divergent pathogens, and are dispensable for bacterial survival [11], [13]. Potential QSI therapeutics fundamentally differ from traditional antimicrobials that are bactericidal and/or bacteriostatic, as they are not expected to disrupt beneficial flora critical for health [51], nor lead to selective resistance [52]. Past approaches to identify QSI compounds against the MvfR-regulon have been predictive, focusing on structural analogues of MvfR-pathway intermediates, and of the MvfR native ligands. Analogues of anthranilic acid, the primary precursor of the HAQs and 2-AA, provided the first demonstration that pharmacological disruption of the MvfR regulon can limit P. aeruginosa pathogenesis [40], [53]. Other compounds were shown to reduce PQS production in vitro, however they lack anti-infective activity [54]–[58]. Similarly, PQS and HHQ analogues reduce a subset of MvfR regulated virulence functions, yet do not provide in vivo efficacy in murine infection models, block bacterial cell persistence, or show activity against multidrug-resistant P. aeruginosa strains [59]–[61]. In addition, P. aeruginosa can modify ligand-based MvfR inhibitors into MvfR activators [62], so drugs based on these compounds could ultimately increase virulence.
In contrast to these predictive approaches, our whole-cell HTS activity-based strategy is not predicated on known QS pathway molecules, and as such, should identify compounds only on their MvfR-regulon QSI activity, independent of structure. Using this strategy and chemical libraries of ∼300,000 small molecular weight compounds, we initially isolated 390 QSIs. We then used a genetic readout system combined with LC/MS to distinguish the inhibitors for robustness and specificity of MvfR pathway inhibition, which validated 17 compounds that belong to 7 divergent chemical families. None of these compounds had been shown to have anti-virulence activity previously, nor proposed as candidates for such. These are the first identified QSIs that inhibit a wide spectrum of MvfR regulon virulence functions, including the pro-acute virulence molecules, HHQ, PQS, and pyocyanin; and the pro-persistent signaling molecule, 2-AA. They are also highly potent, with culture IC50 values of 200–350 nM for HHQ, PQS, and pyocyanin, which is 10–1,000 times more potent than the previous analogue-based inhibitors [60], [62].
Several of the most robust 1st-generation molecules contain the structural backbone of a benzamide and benzimidazole (BB) moiety linked through a thioether bond. These inhibitors demonstrate the validity of our activity-based approach to identify unpredicted chemical structures to develop anti-infective therapeutics. We subsequently used an SAR analysis to define important BB substituents for enhanced QSI activity, including: 1) the aromatic ring, as in M56; 2) the electron-withdrawing substituent at the benzamide para position side-group, as in M34 and M59, especially for pyocyanin production; and 3) the increased bulk of the para substituent on the benzamide moiety of a nitrobenzimidazole derivative, from chlorine to cyano to bromo to iodo, with the M64 phenoxy derivative having the highest activity of all compounds tested. This highly robust 2nd-generation inhibitor notably has significant therapeutic efficacy against both acute and persistent infections in mice, with or without combinatorial antibiotic therapy. In addition, M64 is the first compound identified to inhibit MvfR regulation in divergent P. aeruginosa isolates, including currently untreatable multidrug or pan-resistant strains. Note, this suggests that M64 uptake or action does not appear to be limited in MDR clinical strains that potentially exhibit efflux or influx pumps modifications. Additionally, although M64 contains a nitroaromatic residue, it is not cytotoxic to macrophages (Fig. 6).
Inhibitors of the P. aeruginosa LasR-regulated QS also have in vitro and in vivo anti-virulence potential [11]–[13]. Nonetheless, these reagents may have less practical import and clinical applicability than the MvfR inhibitors, as P. aeruginosa-human isolates often carry lasR mutants. Such mutants may contribute to the fitness of chronic and persistent infections [22], [23], and could cause treatment failure of LasR-specific inhibitors. These lasR cells are likely “cheaters” that benefit from non-mutant cooperators, and do not overcome wild type P. aeruginosa cells in environments lacking selective pressure for LasR activity [52]. In addition, the MvfR pathway product, 2-AA, promotes lasR mutant accumulation [7], so QSI compounds that target MvfR should restrict lasR cheaters from non-mutant cooperators. Moreover, combinatorial therapy of MvfR and LasR inhibitors could enhance target spectrum and clinical potential in acute infections.
Our results show that MvfR is not required for short term bacterial survival in the host, therefore it is not surprising that M64-treated animals do not clear PA14 cells faster than untreated controls in the short term. The higher PA14 clearance observed at day 11 in M64-treated versus not treated animals may suggest a potential fitness advantage for M64-resistant cells for long-term survival in the host. However, we did not observe any M64-resistant cells at day 11 from tissues of animals infected and treated with M64 (data not shown), indicating no clear fitness advantage for M64-resistant mutants in vivo. This is consistent with the fact that no mvfR mutants have been reported in infection sites. Of greater concern are wild-type persister cells, and M64 greatly restricts persister appearance and prevents tissue re-colonization after antibiotic treatment is stopped. Re-colonization may ultimately be responsible for relapsing infection even though, in the setting presented, relapsing infection was not lethal. Transposing these data to a clinical perspective, relapsing infection that occurs after antibiotic treatment arrest may increase patient morbidity/mortality, especially in the case of immunocompromised individuals, and could serve as a source of nosocomial infection as patients remain infected for longer periods of time.
M64 directly binds to MvfR in 1∶1 stoichiometry and inhibits pqs operon expression by reducing MvfR binding to the pqsA promoter, independently of PQS. We propose that M64 induces a non-productive conformational change in MvfR to interrupt effective ligand binding to the native binding domain. Elucidation of the exact M64 binding site and co-crystalization should aid in the design of enhanced anti-MvfR compounds. That M64 does not rescue the survival of macrophages or mice infected with virulence-attenuated mvfR mutant cells further confirms that M64 directly targets MvfR, and its anti-virulence efficacy is not an off-target effect. These results also show that pharmacological in vivo inhibition of MvfR function effectively reduces acute and persistent P. aeruginosa infections. Although M64 and the QS molecule 2-AA [7] both function as MvfR-regulon inhibitors, these two molecules act differently. There are several key differences between 2-AA and M64 : 1) 2-AA promotes the accumulation of AT cells (Fig. 3b and [8]) and host tolerance to infection leading to bacterial persistence [6] whereas M64 prevents these phenotypes (Fig. 3b and 9); 2) 2-AA acts upon at least one of the PQS enzymes of the pqsABCDE operon [7], while M64 does not (Fig. 5a); and 3) M64 is a much more potent MvfR regulon inhibitor than 2-AA as M64 IC50 for the pqsA gene expression inhibition is ∼1,300 times lower (Fig. S7).
M64 exhibits additive effects for P. aeruginosa infections when combined with sub-therapeutic doses of the clinically relevant antibiotic, ciprofloxacin. M64 is also highly efficacious when used in monotherapy in burn and lung acute P. aeruginosa infections in mice. Chronic or persistent infections are often refractory to traditional antibiotics and/or host defense killing mechanisms due to AT and persistent cell subpopulations. Biofilms could form a protective environment for this subpopulation of cells, shielding them from the immune system. The MvfR regulon is reported to control biofilm formation, however in this study the contribution of biofilm or the efficacy of M64 against biofilm formation in our in vivo studies was not assessed. Although many studies have focused on targeting biofilms specifically [63], [64], only very few focused on specifically targeting persister/AT cells. Moreover, current anti-infectives neither target nor limit such cells, leaving a reservoir for re-initiation of infection that underlies chronic/persistent and relapsing infections. AT cells are clinically important, as antibiotics often fail to clear pervasive infections, and the contribution of tolerance to treatment failure and mortality can be as significant as antibiotic resistance. As such, there is considerable need to identify anti-AT compounds that: 1) prevent the formation of AT cells; 2) allow them to be killed; or 3) prevent them from “waking up” when an antibiotic is removed, which can require long-term continuous administration of the anti-infective to assure full clearance. This is both financially costly, and potentially deleterious to the host's natural microbiome. Strategies for killing AT cells include augmentation of antibiotic uptake with sugars [65], stimulation of reactive oxygen species production [66], activation of endogenous proteases [67], or waking-up AT cells with small molecules [68]. Here, M64 prevents AT formation, and in combination with ciprofloxacin, eliminates bacterial rebound and promotes full bacterial clearance. As such, M64 and related compounds provide for the development of robust anti-virulence therapeutics to treat acute, chronic, persistent, and pervasive relapsing infections, in combination with sub-therapeutic levels of traditional antibiotics. In addition, that M64 is efficacious in monotherapy and interferes with AT cell formation, suggests that prophylactic application of M64-based derivatives could reduce antibiotic use. Although M64 will not enter clinical trials directly, it will provide a basis for the development of next generation anti-virulence compounds that contain a similar core structure. As such, it will guide medicinal chemistry efforts to improve solubility and eliminate the potentially problematic nitro group to enhance drug characteristics and ultimately become a new clinical weapon against acute and persistent bacterial infections.
In conclusion, our MvfR-regulon QSI compounds are candidates for the development of next-generation anti-infective/anti-virulence therapeutics, as: 1) they inhibit expression of the MvfR virulence regulon; 2) they do not alter bacterial cell viability or growth; 3) they inhibit the pathogenicity of MDR clinical isolates; 4) they reduce P. aeruginosa virulence in clinically relevant mouse infection models, and compound with antibiotics to block persistent infections; and 5) they restrict the occurrence of AT bacterial cells that underlie chronic and persistent host infections. M64, a 2nd generation inhibitor identified by SAR, is the first identified compound that exhibits significant in vivo therapeutic efficacy against both acute and persistent mammalian infections. Furthermore, that LTTRs regulate virulence regulons in divergent bacterial pathogens [69], and QS functions throughout the eubacteria and archaebacteria, suggest that M64-based anti-infectives could have broad clinical potential against a wide-range of bacterial pathogens.
Materials and Methods
Ethics statement
Animal procedures were performed according to the animal protocols, 2006N000093/2 and 2005N000387/6, approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. The two protocols conform to the USDA Animal Welfare Act, PHS Policy on Humane Care and Use of Laboratory Animals, the “ILAR Guide for the Care and Use of Laboratory Animals” and other applicable laws and regulations.
Bacterial strains, growth conditions, and gene constructs
UCBPP-PA14 (PA14) is a RifR P. aeruginosa human clinical isolate [20]. All mutant strains including mvfR [19] are isogenic to UCBPP-PA14. Unless noted, bacteria were grown at 37°C in LB broth or on LB agar plates containing 75 µg/mL tetracycline, 100 µg/mL rifampicin, and 300 µg/mL carbenicillin,.
mvfR-pPqsABCD bacteria, which have constitutive and MvfR-independent pqs operon expression, were generated by cloning the pqsABCD operon into pDN18 and electroporating this construct into mvfR cells. These bacteria were grown for 6 h in 100 µM of experimental compound, or in 0.2% DMSO as control.
The P. aeruginosa clinical isolates, SH2, Shr12, Shr22, Shr23, Shr33, Shr34, Shr37, Shr40, and Shr42, were obtained from Shriners Hospital, Boston MA.
PpqsA-GFPASV was previously described [38].
The growth kinetics of PA14 WT or PA14 PpqsA-GFP cells were recorded using an automated 96-well plate reader (Sunrise Tecan, Switzerland) at 37°C with 10 s of circular shaking every 15 min, followed by 10 s of settling at which time OD600 nm was detected.
The pqsA-sacB reporter gene was generated by fusing the pqsA promoter to the Bacillus subtilis sacB gene [70]. The pqsA fragment was amplified using PA14 chromosomal DNA and primer pairs 5′GACTAGTCGAGCAAGGGTTGTAACGGTTTTTG3′ and 5′GAAGATCTGACAGAACGTTCCCTCTTCAGCGA3′. The sacB fragment was amplified using pKOBEG-sacB [70] DNA and primer pairs 5′GAAGATCTATGAACATCAAAAAGTTTGCA3′ and 5′AAACTGCAGGTTGATAAGAAATAAAAGAAAATGCC3′. The PpqsA and sacB fragments were digested with SpeI/BglII and BglII/PstI, respectively, and ligated to SpeI/BglII-digested pCTX (TetR). The resultant construct was eletroporated into E. coli SM10 lambda pir and CTX-PpqsA-sacB was integrated into the PA14 chromosome [71]. PA14:CTX-PpqsA-sacB clones were selected on Rif/TetR plates and confirmed by PCR.
HTS
That ligand-bound MvfR binds to and activates the pqsA promoter [16] provided the basis for a biological reporter assay for a high throughput screen (HTS), using PA14 cells carrying a pqsA-sacB reporter gene. pqsA encodes an anthranilate-coenzyme A ligase that activates anthranilic acid and catalyzes the first committed step to HAQ production [72], [73], and is positively regulated by the MvfR protein. sacB encodes levansucrase, which causes toxicity when cells are grown in sucrose, and has been incorporated into allelic exchange vectors to provide counter-selection [74]. Here, the PA14:pqsA-sacB cells die when MvfR activates the pqsA promoter, so compounds that suppress pqsA expression allow growth on sucrose. MvfR inhibition results in reduced sacB expression and viable growth, as determined by absorbance.
Using a plate reader and OD600 nm as readout, the pqsA-sacB construct proved successful in a pilot high-throughput experiment using 4-CABA, an AA analog that effectively inhibits the MvfR regulon [40], as a positive control.
Overnight PA14 pqsA-sacB cultures were subcultured and grown to mid-logarithmic phase, centrifuged, washed, resuspended to a final OD600 nm of 0.05 in LB minus NaCl and plus 10% sucrose, and 30 µl of cells were aliquoted into 384-well plates using a Matrix WellMate. 1.5 mM 4-CABA, a previously described PqsA inhibitor [40], was added to one plate column as the positive control, with another column left compound free for the negative control. 300 nl of a library compound in DMSO was added to each plate via an Epson compound transfer robot, to give a final well concentration of 50 µg/ml. Each library plate was screened in duplicate. After 8 h incubation at 37°C, the OD600 nm was determined for each well using an EnVision plate reader (Perkin-Elmer). The relative inhibition of each compound was from its z-score [75]. This analysis normalizes candidate inhibitory compounds on a plate to plate basis, and corresponds to the standard deviation from the mean plate value.
284,256 compounds in libraries at the Institute of Chemistry and Cell Biology (ICCB)-Longwood screening facility (Fig. S1) were screened in duplicate to identify 532 potential robust inhibitors with strong z-scores. Of these, 390 had limited potential liability, based on their structures. These compounds were tested in a secondary screen at ∼50 µg/mL and ∼25 µg/mL using a reporter construct of the pqsA promoter fused to a short half-life GFP gene [38] as described [7], such that quantitative quenching of fluorescence corresponded to pqsA promoter repression. MvfR inhibition results in reduced GFP expression. This assay eliminated potential false positives, including compounds that negatively affect SacB activity (Fig. S2). Each compound was concomitantly assessed for growth (OD600 nm).
HAQs and pyocyanin quantification
HAQs and 2-AA were quantified in bacterial culture supernatants by LC/MS [16], [76]. Pyocyanin levels were quantified by measuring OD520 nm of chloroform-extracted cultures [77].
Persister cell assay
P. aeruginosa PA14 or mvfR mutant cells were grown with shaking and aeration to mid-logarithmic phase in LB broth, minus and plus exogenous compound. Before antibiotic addition, and as the normalization reference, a culture aliquot was diluted 106 fold in fresh LB (pre-antibiotic) and plated on LB agar for CFU quantification. The remainder of the culture was treated with meropenem to a final concentration of 100×MIC (Minimum Inhibitory Concentration; 10 mg/L) or 5 mg/L amikacin, 0.1 mg/L levofloxacin, or 0.4 mg/L ciprofloxacin. At 16 h post-antibiotic, culture aliquots were washed 2 times in fresh LB to remove antibiotic carry-over, 10-fold serially diluted in LB broth, and plated on LB agar for CFU quantification. This procedure was repeated at 24 h post-AB to ensure that a killing plateau was reached. The persisters fraction was determined as the ratio of normalizers (pre-antibiotic) divided by persisters (24 h post-antibiotic).
Recombinant MvfRc91 purification
The mvfR gene from residue 91 at nucleotides 271 to 273, to the MvfR stop codon at nucleotide 999, plus 129 bp downstream, was cloned into the Nde1 and Xho1 sites of pET16B, to generate pET16B-MvfRc91. E. coli BL21(DE3) cells harboring pET16b-MvfRc91 were grown at 37°C to OD600 nm 0.6 [16]. His-tagged MvfRc91 expression was induced with 0.5 mM IPTG at 20°C for 16 h, and the cells were harvested by centrifugation. The bacterial pellet was resuspended in Tris buffer (20 mM Tris-HCl, 300 mM NaCl, pH8.0) with 10 mM imidazole, and lysed by sonication. The soluble fraction was collected by centrifugation and filtration, and separated on a Ni-NTA column equilibrated with Tris buffer containing 50 mM imidazole. After column wash, the His-tagged MvfRc91 protein was eluted with a 0.1 M–1.0 M imidazole gradient. The MvfRc91fractions were pooled and dialyzed in phosphate buffer (pH 8.0) with 300 mM NaCl and 2.5 mM β-mercaptoethanol.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as described in [78] using VSV-G-tagged MvfR in PA14 cells. To construct the MvfR - VSV-G integration vector pP30ΔFRT-MvfR – VSV-G, the 316 bp fragment corresponding to mvfR 630–945 region was amplified by PCR using forward primer mvfR HindIII (5′-GACGTAAGCTTGGTCAGCGACAAGGTGCTCTTC-3′) and reverse primer mvfR_NotI (5′-GAAATGCGGCCGCCTGCACCGTTTCGACGATGCTCGG-3′). The PCR product was then treated with HindIII and NotI and cloned into the HindIII and NotI sites of pP30ΔFRT [79]. PA14 expressing MvfR-VSV-G was obtained via conjugation of PA14 with E. coli S17-1 λpir carrying the pP30ΔFRT-MvfR - VSV-G plasmid. Conjugants with chromosomally-integrated plasmid were selected on LB plate containing 30 µg/ml gentamicin. Plasmid backbone excision was performed by transforming plasmid pFLP2, which encodes FLP recombinase. Expression of the MvfR – VSV-G protein was confirmed by western blot analysis using a rabbit anti – VSV-G primary antibody (Sigma–Aldrich) and a goat anti-rabbit HRP-conjugated secondary antibody (GE Healthcare). Loading control was performed for housekeeping protein RpoD detected with a mouse anti-RpoD primary antibody (Neoclone) and a sheep anti-mouse HRP-conjugated secondary antibody (GE Healthcare Science).
Binding of tagged MvfR protein to the non-MvfR-regulated rpoD promoter DNA was used as the negative control. 5 ml culture aliquots were inoculated at OD600 nm 0.03, and grown at 37°C to OD600 nm 0.75, minus and plus 0.24 µM M64, and minus and plus 38 µM PQS. Cross-linking and ChIP were as described [78]. Quantitative PCR used oligonucleotides to the pqs operon, and to the rpoD promoter as negative control [31], [80]. MvfR binding was expressed as the percent of total input DNA. Data were averaged from at least 3 replicates.
Isothermal titration calorimetry (ITC)
ITC experiments were performed using a VP-ITC (Microcal). Ligand and protein were in 100 mM phosphate buffer (pH 8.0) plus 2.5 mM β-mercaptoethanol and 10% methanol. For individual titrations, 10 µl of 200 µM M64 was injected using a computer-controlled microsyringe at 180 second intervals into 1.5 ml of 19 µM MvfRc91, with the sample cell stirred at 300 rpm, and the heat produced was measured at 25°C. The heat originating from M64 injection into buffer alone was subtracted from the raw data. The dissociation constant was calculated using Origin Software (Microcal) by plotting heat per injection (µJ) versus the titrated MvfRc91/M64 stoichiometry.
Macrophage cytotoxicity
PA14 or mvfR bacterial cultures were grown to OD600 nm 2.0, minus and plus 50 µM experimental compound. Raw264.7 macrophage cells were cultured in Dulbecco's modified Eagles medium (DMEM) containing 10% FCS, 2 mM glutamine, and antibiotic-antimycotic; washed with PBS (Mg2+ and Ca2+ free); resuspended in antibiotic-free medium; and infected with 100 MOI of bacteria. Following 3 h incubation at 37°C under 5% CO2, macrophages were washed and incubated for a further 3 h with DMEM containing polymixin B and gentamycin to kill extracellular bacteria. Raw264.7 viability was assessed using the MTT (3-[4, 5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2H-tetrazolium bromide) assay for early detection of eukaryotic cell death [81]. Briefly, macrophages were incubated in 200 µl PBS containing 200 µg/ml MTT (Sigma-Aldrich) in a 96-well culture plate for 2 h at 37°C under 5% CO2, and the dissolved MTT was converted to insoluble purple formazan via intracellular mitochondrial activity. The supernatant was removed and the cells were lysed for 10 min with 95% isopropanol and 5% formic acid. Converted dye absorbance was measured at OD570 nm, with OD690 nm as the reference wavelength, in a Sunrise plate reader (Tecan Group Ltd, Männedorf, Switzerland). Per cent infected cell viability was calculated by dividing the OD570 nm of infected versus uninfected culture.
Murine burn and infection model to address anti-virulence and anti-persistence compound efficacy
A murine thermal injury model was used to assess bacterial pathogenicity in 6–7 wk-old CD-1 mice, as described [20]. Briefly, animals were anesthetized with Xylazine (13 mg/kg, i.p.) and Ketamine (87 mg/kg, i.p.), thermally injured (5–8% of body surface) on the shaved abdomen dermis, and intra-dermally infected into the burn eschar.
To assess MvfR inhibitory compound activity in acute infection, animals were inoculated with 5×104 PA14 cells in 100 µl of 10 mM MgSO4; and injected IV into the tail vein with M64 (4 mg/kg in 15% cremophore) and/or ciprofloxacin (10 mg/kg or 0.4 mg/kg), twice a day for up to 6 or 4 days post-infection, respectively, for M64 or ciprofloxacin. Mice survival was assessed over the course of 7 or 14 days, with 10 animals per experimental group, and CFUs in adjacent or underlying muscle were quantified at 2, 3, 4, 6, 8, 11, and 14 days post-infection, as described [7]. Samples from underlying muscle represent assessment of bacterial CFUs at the site of inoculation, whereas the adjacent muscle samples provide a read out of the bacterial dissemination from the site of inoculation. A sub-therapeutic concentration of ciprofloxacin (0.4 mg/kg) was used to assess M64 and ciprofloxacin additive effect. Kaplan-Meier statistical analysis was performed using Prism Graphpad software.
To assess MvfR-regulon inhibitor efficacy in persistent infections, persistent nonlethal infections were produced in burned mice by using a lower bacterial inoculum than the one used above. Mice were inoculated with ∼6×103 PA14 cells in 100 µl of 10 mM MgSO4. Ciprofloxacin (10 mg/kg) plus or minus, M64 (4 mg/kg in 15% cremophore) were IV injected into the tail vein twice a day for up to 6 or 4 days post-infection for M64 or ciprofloxacin, respectively. CFUs in adjacent or underlying muscle were quantified at 2, 3, 4, 6, 8, 11, and 14 days post-infection, as described [7]. MIC was calculated via the multivariate E-Test (Biomerieux) [7].
Mouse lung infection model
M64 in vivo efficacy was further assessed in a murine lung acute infection model [82]. 6 wk-old CD-1 mice were anaesthetized with Xylazine (13 mg/kg, IP) and Ketamine (87 mg/kg, IP), and intranasally inoculated with 20 µL of 5×106 PA14 or 8×106 mvfR mutant cells. M64 (4 mg/kg or 12 mg/kg) was injected IV at 2, 4, 8, and 12 h, followed by injections twice daily through 4 days post-infection. Animals were held in a vertical position for 3–5 min to facilitate distal alveolar migration of the bacteria by gravity. Mice survival was assessed over 6 days, with 10 or more animals per experimental group. Kaplan-Meier statistical analysis was performed using Prism Graphpad software.
In vivo molecular MR Imaging to address anti-virulence compound efficacy
In vivo molecular magnetic resonance imaging positive contrast method exploits the chemical shift induced by ultra-small super-paramagnetic iron oxide (USPIO) nanoparticles, known generically as Ferumoxtran-10 commercially and as Combidex in the U.S. (Advanced Magnetics, Cambridge, MA). We used the USPIO nanoparticles as the molecular imaging MRI contrast agent. Six weeks old CD-1 mice were anesthetized with Xylazine (13 mg/kg, IP) and Ketamine (87 mg/kg, IP) and a leg thermal injury of ∼8% total burn surface area was produced on the right thigh muscle. Six hours post-burn and infection 500 mg of Ferumextron-10 suspension was injected by intravenous injection in the tail vein. Mice were randomized into one experimental and one control group (N = 6 per group). The experimental group consisted of burned and infected mice, injected with USPIO and injected with the anti-infective compound M64. The control group consisted of burned and infected mice injected with USPIO. The mice were imaged 12 hour post-burn and infection. We imaged the accumulation of USPIO-labeled macrophages at the P. aeruginosa infection site in the mouse burn and infection model. Briefly, imaging was performed in a 4.7 T horizontal magnet (20 cm bore, Bruker Avance console) equipped with a 39 G/cm gradient system, using a custom volume coil of 3 cm inner diameter and 10 cm active length. This set-up permits high B1 for extended periods of time necessary for T2ρ and provides extended homogeneity. Imaging was performed with RARE acceleration factor two. The ORI-T2ρ sequence used a spin-locking pulse block for relaxation in the rotating frame between the 90° and 180° RF pulses of the RARE sequence. Magnetization inversion was achieved with 180° adiabatic full passage pulses using HS4 adiabatic pulses (3 ms pulse duration, BW = 7 kHz) [83]. The spin-locking block was implemented with the MLEV-4 scheme (12 ms duration of spin-lock) [84]. Water and fat were suppressed using frequency-selective ten-lobed sinc pulses (400 Hz pulse bandwidth for water, 800 Hz for fat), followed by spoiling gradients to dephase the transverse magnetization. Typical parameters were RARE acceleration factor 2, effective echo time (TE) 9.93 ms, repetition time TR 2240 ms, with 8 averages. Anatomical reference images were acquired with RARE or proton-density weighted FLASH (fast-low angle shot) imaging. Negative contrast was achieved with a series of FLASH images with increasing echo time for T2* weighting, with typical values α = 35°, TR = 500 ms, TE = 4, 6, 8, 12, and 14 ms. The same slice prescription was used for all sequences. Typically, 10 axial slices were acquired in the burned region (1 mm thickness, 1.5 mm gap, 3×3 cm FOV, 128×128 matrix size, 8 averages). Typical MR imaging times were 1.3 hr per animal.
MIC determination and antibiotic sensitivity/resistance profiling
PA14 MIC for antibiotic treatment was as described [85]. Antibiotic sensitivity/resistance profiles were determined using the disc diffusion method [86].
Statistical analysis
Data from 3 or more independent experiments were analyzed using the Student's t-test or one way ANOVA with Dunnett's post-test when required, and animal data were analyzed using the Kaplan-Meier survivability test. P values<0.05, <0.01, and <0.001 were considered statistically significant, very significant, and highly significant, respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BoucherHW, TalbotGH, BradleyJS, EdwardsJE, GilbertD, et al. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48 : 1–12.
2. LivermoreDM (2012) Fourteen years in resistance. Int J Antimicrob Agents 39 : 283–294.
3. SpellbergB, GuidosR, GilbertD, BradleyJ, BoucherHW, et al. (2008) The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46 : 155–164.
4. ParkerCT, SperandioV (2009) Cell-to-cell signalling during pathogenesis. Cell Microbiol 11 : 363–369.
5. NgWL, BasslerBL (2009) Bacterial quorum-sensing network architectures. Annu Rev Genet 43 : 197–222.
6. BandyopadhayaA, KesarwaniM, QueYA, HeJ, PadfieldK, et al. (2012) The quorum sensing volatile molecule 2-amino acetophenon modulates host immune responses in a manner that promotes life with unwanted guests. PLoS Pathog 8: e1003024.
7. KesarwaniM, HazanR, HeJ, QueY, ApidianakisY, et al. (2011) A Quorum Sensing Regulated Small Volatile Molecule Reduces Acute Virulence and Promotes Chronic Infection Phenotypes. PLoS Pathogens 7: e1002192.
8. QueYA, HazanR, StrobelB, MauraD, HeJ, et al. (2013) A quorum sensing small volatile molecule promotes antibiotic tolerance in bacteria. PLoS One 8: e80140.
9. MökerN, DeanCR, TaoJ (2010) Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. Journal of bacteriology 192 : 1946–1955.
10. VegaNM, AllisonKR, KhalilAS, CollinsJJ (2012) Signaling-mediated bacterial persister formation. Nat Chem Biol 8 : 431–433.
11. RaskoDA, SperandioV (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 9 : 117–128.
12. DefoirdtT, BrackmanG, CoenyeT (2013) Quorum sensing inhibitors: how strong is the evidence? Trends Microbiol 21 : 619–624.
13. NjorogeJ, SperandioV (2009) Jamming bacterial communication: new approaches for the treatment of infectious diseases. EMBO Mol Med 1 : 201–210.
14. WilliamsP, CamaraM (2009) Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol 12 : 182–191.
15. DezielE, LepineF, MilotS, HeJ, MindrinosMN, et al. (2004) Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A 101 : 1339–1344.
16. XiaoG, DezielE, HeJ, LepineF, LesicB, et al. (2006) MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol Microbiol 62 : 1689–1699.
17. SchellMA (1993) Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol 47 : 597–626.
18. TangHB, DiMangoE, BryanR, GambelloM, IglewskiBH, et al. (1996) Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun 64 : 37–43.
19. CaoH, KrishnanG, GoumnerovB, TsongalisJ, TompkinsR, et al. (2001) A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proceedings of the National Academy of Sciences 98 : 14613.
20. RahmeLG, StevensEJ, WolfortSF, ShaoJ, TompkinsRG, et al. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science (New York, NY) 268 : 1899–1902.
21. PearsonJP, PesciEC, IglewskiBH (1997) Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol 179 : 5756–5767.
22. D'ArgenioDA, WuM, HoffmanLR, KulasekaraHD, DezielE, et al. (2007) Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol 64 : 512–533.
23. SmithEE, BuckleyDG, WuZ, SaenphimmachakC, HoffmanLR, et al. (2006) Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103 : 8487–8492.
24. LauGW, GoumnerovBC, WalendziewiczCL, HewitsonJ, XiaoW, et al. (2003) The Drosophila melanogaster toll pathway participates in resistance to infection by the gram-negative human pathogen Pseudomonas aeruginosa. Infect Immun 71 : 4059–4066.
25. DézielE, GopalanS, TampakakiAP, LépineF, PadfieldKE, et al. (2005) The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol Microbiol 55 : 998–1014.
26. WadeDS, CalfeeMW, RochaER, LingEA, EngstromE, et al. (2005) Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J Bacteriol 187 : 4372–4380.
27. QueY, HazanR, RyanCM, MilotS, LépineF, et al. (2011) Production of Pseudomonas aeruginosa Intercellular Small Signaling Molecules in Human Burn Wounds. Journal of Pathogens 2011 : 1–5.
28. TaylorGW, MachanZA, MehmetS, ColePJ, WilsonR (1995) Rapid identification of 4-hydroxy-2-alkylquinolines produced by Pseudomonas aeruginosa using gas chromatography-electron-capture mass spectrometry. J Chromatogr B Biomed Appl 664 : 458–462.
29. DiggleSP, MatthijsS, WrightVJ, FletcherMP, ChhabraSR, et al. (2007) The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chemistry & biology 14 : 87–96.
30. HeebS, FletcherMP, ChhabraSR, DiggleSP, WilliamsP, et al. (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35 : 247–274.
31. HazanR, HeJ, XiaoG, DekimpeV, ApidianakisY, et al. (2010) Homeostatic interplay between bacterial cell-cell signaling and iron in virulence. PLoS Pathogens 6: e1000810.
32. Scott-ThomasAJ, SyhreM, PattemorePK, EptonM, LaingR, et al. (2010) 2-Aminoacetophenone as a potential breath biomarker for Pseudomonas aeruginosa in the cystic fibrosis lung. BMC Pulm Med 10 : 56.
33. TzikaAA, ConstantinouC, BandyopadhayaA, PsychogiosN, LeeS, et al. (2013) A small volatile bacterial molecule triggers mitochondrial dysfunction in murine skeletal muscle. PLoS One 8: e74528.
34. LewisK (2010) Persister Cells. Annual review of microbiology 64 : 357–72.
35. FauvartM, De GrooteVN, MichielsJ (2011) Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60 : 699–709.
36. GefenO, BalabanNQ (2009) The importance of being persistent: heterogeneity of bacterial populations under antibiotic stress. FEMS microbiology reviews 33 : 704–717.
37. PelicicV, ReyratJM, GicquelB (1996) Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on mycobacteria. J Bacteriol 178 : 1197–1199.
38. YangL, BarkenKB, SkindersoeME, ChristensenAB, GivskovM, et al. (2007) Effects of iron on DNA release and biofilm development by Pseudomonas aeruginosa. Microbiology (Reading, England) 153 : 1318–1328.
39. FarrowJM, PesciEC (2007) Two distinct pathways supply anthranilate as a precursor of the Pseudomonas quinolone signal. Journal of bacteriology 189 : 3425–3433.
40. LesicB, LepineF, DezielE, ZhangJ, ZhangQ, et al. (2007) Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog 3 : 1229–1239.
41. HazanR, QueYA, MauraD, RahmeLG (2012) A method for high throughput determination of viable bacteria cell counts in 96-well plates. BMC Microbiol 12 : 259.
42. LepineF, DekimpeV, LesicB, MilotS, LesimpleA, et al. (2007) PqsA is required for the biosynthesis of 2,4-dihydroxyquinoline (DHQ), a newly identified metabolite produced by Pseudomonas aeruginosa and Burkholderia thailandensis. Biol Chem 388 : 839–845.
43. ZhangYM, FrankMW, ZhuK, MayasundariA, RockCO (2008) PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J Biol Chem 283 : 28788–28794.
44. XiaoG, HeJ, RahmeLG (2006) Mutation analysis of the Pseudomonas aeruginosa mvfR and pqsABCDE gene promoters demonstrates complex quorum-sensing circuitry. Microbiology 152 : 1679–1686.
45. RahmeLG, AusubelFM, CaoH, DrenkardE, GoumnerovBC, et al. (2000) Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci U S A 97 : 8815–8821.
46. RahmeLG, TanMW, LeL, WongSM, TompkinsRG, et al. (1997) Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc Natl Acad Sci U S A 94 : 13245–13250.
47. Mahajan-MiklosS, TanMW, RahmeLG, AusubelFM (1999) Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96 : 47–56.
48. LavoieEG, WangdiT, KazmierczakBI (2011) Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect 13 : 1133–1145.
49. BjarnsholtT, Kirketerp-MollerK, JensenPO, MadsenKG, PhippsR, et al. (2008) Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen 16 : 2–10.
50. MulcahyLR, BurnsJL, LoryS, LewisK (2010) Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. Journal of bacteriology 192 : 6191–6199.
51. UbedaC, PamerEG (2012) Antibiotics, microbiota, and immune defense. Trends Immunol 33 : 459–466.
52. MellbyeB, SchusterM (2011) The sociomicrobiology of antivirulence drug resistance: a proof of concept. MBio 2: e00131–11.
53. CalfeeMW, ColemanJP, PesciEC (2001) Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98 : 11633–11637.
54. LeeJH, KimYG, ChoMH, KimJA, LeeJ (2012) 7-fluoroindole as an antivirulence compound against Pseudomonas aeruginosa. FEMS Microbiol Lett 329 : 36–44.
55. TashiroY, ToyofukuM, Nakajima-KambeT, UchiyamaH, NomuraN (2010) Bicyclic compounds repress membrane vesicle production and Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. FEMS Microbiol Lett 304 : 123–130.
56. YangL, LiuY, SternbergC, MolinS (2010) Evaluation of enoyl-acyl carrier protein reductase inhibitors as Pseudomonas aeruginosa quorum-quenching reagents. Molecules 15 : 780–792.
57. YangL, RybtkeMT, JakobsenTH, HentzerM, BjarnsholtT, et al. (2009) Computer-aided identification of recognized drugs as Pseudomonas aeruginosa quorum-sensing inhibitors. Antimicrob Agents Chemother 53 : 2432–2443.
58. CuginiC, CalfeeMW, FarrowJM3rd, MoralesDK, PesciEC, et al. (2007) Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol Microbiol 65 : 896–906.
59. LuC, KirschB, ZimmerC, de JongJC, HennC, et al. (2012) Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem Biol 19 : 381–390.
60. IlangovanA, FletcherM, RampioniG, PustelnyC, RumbaughK, et al. (2013) Structural Basis for Native Agonist and Synthetic Inhibitor Recognition by the Pseudomonas aeruginosa Quorum Sensing Regulator PqsR (MvfR). PLoS Pathog 9: e1003508 doi:1003510.1001371/journal.ppat.1003508
61. KleinT, HennC, de JongJC, ZimmerC, KirschB, et al. (2012) Identification of small-molecule antagonists of the Pseudomonas aeruginosa transcriptional regulator PqsR: biophysically guided hit discovery and optimization. ACS Chem Biol 7 : 1496–1501.
62. LuC, MaurerCK, KirschB, SteinbachA, HartmannRW (2013) Overcoming the Unexpected Functional Inversion of a PqsR Antagonist in Pseudomonas aeruginosa: An In Vivo Potent Antivirulence Agent Targeting pqs Quorum Sensing. Angew Chem Int Ed Engl 126 : 1127–1130.
63. ChungPY, TohYS (2014) Anti-biofilm agents: recent breakthrough against multi-drug resistant Staphylococcus aureus. Pathog Dis 70 : 231–239.
64. BjarnsholtT, CiofuO, MolinS, GivskovM, HoibyN (2013) Applying insights from biofilm biology to drug development - can a new approach be developed? Nat Rev Drug Discov 12 : 791–808.
65. AllisonKR, BrynildsenMP, CollinsJJ (2011) Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473 : 216–220.
66. GrantSS, KaufmannBB, ChandNS, HaseleyN, HungDT (2012) Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci U S A 109 : 12147–12152.
67. ConlonBP, NakayasuES, FleckLE, LaFleurMD, IsabellaVM, et al. (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503 : 365–370.
68. KimJS, HeoP, YangTJ, LeeKS, JinYS, et al. (2011) Bacterial persisters tolerate antibiotics by not producing hydroxyl radicals. Biochem Biophys Res Commun 413 : 105–110.
69. KrushkalJ, QuY, LovleyDR, AdkinsRM (2012) Phylogenetic classification of diverse LysR-type transcriptional regulators of a model prokaryote Geobacter sulfurreducens. J Mol Evol 74 : 187–205.
70. DerbiseA, LesicB, DacheuxD, GhigoJM, CarnielE (2003) A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol Med Microbiol 38 : 113–116.
71. LesicB, RahmeLG (2008) Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol Biol 9 : 20.
72. ColemanJP, HudsonLL, McKnightSL, FarrowJM, CalfeeMW, et al. (2008) Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. Journal of bacteriology 190 : 1247–1255.
73. GallagherLA, McKnightSL, KuznetsovaMS, PesciEC, ManoilC (2002) Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. Journal of bacteriology 184 : 6472.
74. PelicicV, ReyratJM, GicquelB (1996) Generation of unmarked directed mutations in mycobacteria, using sucrose counter-selectable suicide vectors. Mol Microbiol 20 : 919–925.
75. ZhangJH, ChungTD, OldenburgKR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4 : 67–73.
76. LepineF, DezielE, MilotS, RahmeLG (2003) A stable isotope dilution assay for the quantification of the Pseudomonas quinolone signal in Pseudomonas aeruginosa cultures. Biochim Biophys Acta 1622 : 36–41.
77. EssarDW, EberlyL, HaderoA, CrawfordIP (1990) Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J Bacteriol 172 : 884–900.
78. CastangS, McManusHR, TurnerKH, DoveSL (2008) H-NS family members function coordinately in an opportunistic pathogen. Proc Natl Acad Sci U S A 105 : 18947–18952.
79. Vallet-GelyI, DonovanKE, FangR, JoungJK, DoveSL (2005) Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102 : 11082–11087.
80. SavliH, KaradenizliA, KolayliF, GundesS, OzbekU, et al. (2003) Expression stability of six housekeeping genes: A proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J Med Microbiol 52 : 403–408.
81. MosmannT (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65 : 55–63.
82. ComolliJC, HauserAR, WaiteL, WhitchurchCB, MattickJS, et al. (1999) Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun 67 : 3625–3630.
83. GarwoodM, DelaBarreL (2001) The return of the frequency sweep: designing adiabatic pulses for contemporary NMR. J Magn Reson 153 : 155–177.
84. LevittMH, FreemanR, FrenkielT (1982) Broadband heteronuclear decoupling. J Magn Reson 47 : 328–330.
85. Rankin ID (2005) MIC testing. In: Coyle MB, editor. Manual of Antimicrobial Susceptibility Testing: American Society for Microbiology. pp. 53–62.
86. BauerAW, RobertsCEJr, KirbyWM (1959) Single disc versus multiple disc and plate dilution techniques for antibiotic sensitivity testing. Antibiot Annu 7 : 574–580.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Disruption of Fas-Fas Ligand Signaling, Apoptosis, and Innate Immunity by Bacterial Pathogens
- Ly6C Monocyte Recruitment Is Responsible for Th2 Associated Host-Protective Macrophage Accumulation in Liver Inflammation due to Schistosomiasis
- Host Responses to Group A Streptococcus: Cell Death and Inflammation
- Pathogenicity and Epithelial Immunity