
Phenotypic Plasticity of the Drosophila Transcriptome
Phenotypic plasticity is the ability of a single genotype to produce different phenotypes in response to changing environments. We assessed variation in genome-wide gene expression and four fitness-related phenotypes of an outbred Drosophila melanogaster population under 20 different physiological, social, nutritional, chemical, and physical environments; and we compared the phenotypically plastic transcripts to genetically variable transcripts in a single environment. The environmentally sensitive transcriptome consists of two transcript categories, which comprise ∼15% of expressed transcripts. Class I transcripts are genetically variable and associated with detoxification, metabolism, proteolysis, heat shock proteins, and transcriptional regulation. Class II transcripts have low genetic variance and show sexually dimorphic expression enriched for reproductive functions. Clustering analysis of Class I transcripts reveals a fragmented modular organization and distinct environmentally responsive transcriptional signatures for the four fitness-related traits. Our analysis suggests that a restricted environmentally responsive segment of the transcriptome preserves the balance between phenotypic plasticity and environmental canalization.
Published in the journal:
Phenotypic Plasticity of the Drosophila Transcriptome. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002593
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002593
Summary
Phenotypic plasticity is the ability of a single genotype to produce different phenotypes in response to changing environments. We assessed variation in genome-wide gene expression and four fitness-related phenotypes of an outbred Drosophila melanogaster population under 20 different physiological, social, nutritional, chemical, and physical environments; and we compared the phenotypically plastic transcripts to genetically variable transcripts in a single environment. The environmentally sensitive transcriptome consists of two transcript categories, which comprise ∼15% of expressed transcripts. Class I transcripts are genetically variable and associated with detoxification, metabolism, proteolysis, heat shock proteins, and transcriptional regulation. Class II transcripts have low genetic variance and show sexually dimorphic expression enriched for reproductive functions. Clustering analysis of Class I transcripts reveals a fragmented modular organization and distinct environmentally responsive transcriptional signatures for the four fitness-related traits. Our analysis suggests that a restricted environmentally responsive segment of the transcriptome preserves the balance between phenotypic plasticity and environmental canalization.
Introduction
Phenotypic plasticity is the ability of a single genotype to give rise to different phenotypes in different environments [1]. Phenotypic plasticity is the counterpoint to environmental canalization [2]–[3], whereby genotypes produce the same phenotype in different environments. Phenotypic plasticity allows organisms to respond rapidly to changing environmental conditions without the time lag required for response to natural selection on segregating allelic variants and without the cost of selection, while environmental canalization buffers phenotypes against environmental perturbations. The balance between plasticity and robustness is thus crucial for optimal fitness [3]–[4] in variable environments, but the genetic basis for phenotypic plasticity has remained poorly defined.
Elucidating the genetic underpinnings of phenotypic plasticity (and its converse, environmental canalization) requires that we determine what fraction of the genome is environmentally sensitive, which genes respond to the same or different environmental perturbations and how expression of environmentally sensitive genes is correlated with plasticity of organismal phenotypes. It is also necessary to determine what the relationship is between genetic variance and phenotypic plasticity, whether the same genes affecting phenotypic plasticity for a trait also affect genetic variation for that trait, and whether environmentally plastic and environmentally robust genes evolve at different rates. Although previous studies have analyzed changes in gene expression under one or few different environmental or physiological conditions [5]–[11], the study presented here is the first comprehensive study that analyzes co-variation among environmentally responsive genes across a wide range of environments in a defined outbred population reconstructed from inbred lines with documented expression profiles, enabling us to compare genotypic and environmental variation.
We examined phenotypic plasticity in genome-wide gene expression and four organismal phenotypes related to reproductive fitness in a population generated by crossing 40 wild-derived inbred D. melanogaster lines [12]. The majority of the transcriptome shows robust expression across a range of environmental challenges, including different nutritional rearing conditions, physical stress conditions, chemical exposures, social crowding during larval or adult stages, and aging. Approximately 15% of transcripts are phenotypically plastic. By comparing genotypic variation among the original 40 wild-derived inbred lines under standard growth conditions, documented earlier [12], with environmental variation of transcript abundance levels in the reconstituted outbred population, we were able to discriminate two distinct classes of environmentally responsive transcripts, which we have designated Class I and Class II transcripts.
Results
Phenotypic Plasticity of the Transcriptome
To identify phenotypically plastic and environmentally canalized transcripts, we assessed genome-wide gene expression of flies exposed to 20 treatments, including a control treatment of mated flies reared under standard conditions, and different nutrient or drug supplements, exposure to different physical and social environments, and maintenance at different reproductive states. Of the 18,800 transcripts represented on the microarray, 14,400 (76.6%) generated signal intensities above background under at least one treatment, similar to the proportion of the transcriptome detected in a previous study, in which transcript profiles were obtained separately for the 40 individual genotypes that gave rise to our outbred population [12]. Analysis of variance of microarray intensity signals across all 20 rearing conditions revealed 1,249 transcripts that showed a significant treatment effect (8.7%), 6,745 transcripts that showed a sex effect (46.8%), and 200 transcripts with a significant treatment by sex interaction term (1.4%) at a false discovery rate of 0.05. Thus, the majority of the transcriptome is remarkably robust and buffered against diverse environmental challenges.
We refer to the 1,249 transcripts exhibiting phenotypic plasticity as quantified by the significant treatment term in the ANOVAs as Class I transcripts. To simplify statistical analyses and maintain optimum power we excluded 166 Class I transcripts that also had significant treatment by sex interaction terms, giving 1,133 phenotypically plastic transcripts for further analyses (Table S1). We grouped these transcripts according to their relative expression levels across the 20 conditions (Figure S1). The highest relative gene expression levels were observed in flies exposed to low temperature, dopamine, nicotine or high sugar, and the lowest relative levels in flies exposed to heat shock or grown on high yeast medium. Surprisingly, overall relative gene expression levels are either uniformly higher or lower (more than 70% show higher abundance levels than the median) across the 20 conditions; however, starvation stress resistance, aging, larval crowding and exposure to high temperature, result in substantial variation among relative expression levels (Figure S1).
To further examine the relationship between gene expression and environmental exposure, we compared transcript abundance levels of the Class I transcripts under the different treatments to the standard rearing condition with post hoc least square difference (LSD) tests (p<0.05; Table S2). Heat shock has the greatest impact on gene expression (589 transcripts), whereas fluoxetine changes expression of only four transcripts (Figure 1A). Many transcripts show altered expression under multiple treatments; for example, 167 transcripts show altered expression both after heat shock and exposure to starvation (Figure 1A). The majority of transcripts do not undergo significant changes compared to the standard condition (Figure 1B).

Among the 1,133 Class I transcripts, 691 are computationally predicted with unknown function, 14 probe sets correspond to intergenic regions, non-coding RNAs and transposons, and 428 are annotated. The transcripts that show altered expression after heat shock include 13 Heat shock proteins (Hsps), 60 proteases, 17 members of the cytochrome P450 family (Cyps), two glutathione-S-transferases (Gsts), six UDP-glucose glycoprotein glycosyltransferases (Ugts), and six immune-induced molecules (IM) (Figure 2, Table S2; Figure S2). A variety of additional transcripts in diverse gene ontology (GO) categories also respond to environmental challenges (Table S3). Nine of the 13 Hsps upregulated after heat shock are also upregulated after induction of chill coma (Figure S2). The abundance of heat shock proteins and proteases encoded by environmentally sensitive genes reflect mechanisms for environmental adaptation of the proteome. Heat shock proteins may offer protection for nascent polypeptides under adverse temperature conditions, while one function of environmentally sensitive proteases may be to facilitate de novo protein synthesis by providing a pool of amino acids through degradation of dispensable proteins.

Modules of Phenotypically Plastic Transcripts
We asked to what extent expression patterns of phenotypically plastic transcripts are co-regulated across environmental treatments. A previous study on the 40 inbred lines from which our outbred population is derived demonstrated that the genetically variable transcriptome (10,096 transcripts) is highly inter-correlated and can be subdivided into 241 co-regulated modules [12], identified by modulated modularity clustering (MMC) [12]–[13]. Here, we used MMC to identify transcripts that covary across different treatments (Figure 3A, Table S1). This analysis partitioned the 1,133 Class I transcripts into 103 small, but highly correlated transcriptional modules (the average absolute correlation coefficient, |r|, within modules is at least 0.56). Extensive cross-module correlations are also evident. Negative correlations are rare, in agreement with the overall uniform up - or down-regulation of transcripts (Figure S1). All seven IM transcripts group together in module 71. A putative IM, CG15065, which is genetically correlated with IM1 and IM3 [12], is also contained in this module (Figure S2). These results show that changes in environmental conditions can cause fragmentation of the highly intercorrelated structure of the transcriptome observed under the standard growth condition [12].

Phenotypically Plastic Transcription Factors
What are the cellular mechanisms that regulate transcriptional responses to environmental changes? As a first step to investigating how environmental stimuli may influence transcriptional regulation, we asked which transcription factors show altered expression under the different environmental conditions. Among the Class I transcripts, we identified 26 transcripts that encode transcriptional regulators, of which 25 were differentially expressed relative to the standard growth condition (Figure 3B, Figure 4A). Many of these transcription factors occur together in the same transcriptional modules (Figure 3B). The complexity of the interrelationships between transcript abundance levels of different transcription factors under different growth conditions is further illustrated in Figure 4A.

Each transcription factor can exert wide-ranging effects on networks of interacting genes that include other regulatory genes, non-regulatory genes and miRNAs [14]–[15] (Figure 4B). Direct genetic, protein-protein and gene-protein interactions among these phenotypically plastic transcription factors appear compartmentalized, with little overlap between interacting components related to each transcription factor. In contrast, there is an extensive network of interactions among microRNAs and different transcription factors (Figure 4B). Extensive interactions of transcription factors with miRNAs suggest that these may also contribute to phenotypic plasticity of the transcriptome [14]–[15]. In addition, seven long non-coding RNAs and unannotated intergenic regions are phenotypically plastic, and genes that encode several phenotypically plastic transcripts contain overlapping or flanking sequences for short non-coding RNAs (Table S1). Finally, we note that predicted transcripts of unknown function may also play a regulatory role. In addition, transcriptional regulators that themselves show no change in gene expression may be regulated by phenotypically plastic post-translational modifications.
Phenotypic Plasticity of Organismal Phenotypes
We next asked to what extent the phenotypic plasticity in gene expression is associated with phenotypic plasticity of organismal phenotypes. We assessed phenotypic plasticity of four fitness-related phenotypes: development time, lifespan, starvation stress resistance, and chill coma recovery time.
Development is exquisitely sensitive to environmental conditions [16]–[17] (Figure 5A) and is accelerated when flies are grown on medium supplemented with high yeast, and delayed when the medium is supplemented with high sugar. When grown on both high sugar and high yeast, development time is identical to that under the standard growth condition. Growth at 28°C also accelerates development, but reduces survival, whereas growth at 18°C delays development about 2-fold. Larval crowding and exposure to the chemical oxidative stress agent menadione sodium bisulfite results in delayed development along with reduced survival. Medium supplemented with 10% ethanol has a small effect on development time and survival. All other treatments lead to slower development compared to the standard condition.

In addition to prolonging development, growth at 18°C results in a two-fold increase in lifespan (Figure 5B). Furthermore, virgin females live longer than mated females, as expected [18]. When reared under standard conditions, subsequent food deprivation allows females to survive twice as long as males (Figure 5C). Survival curves indicate a trend towards longer survival times for both sexes when flies are maintained at high density, either as larvae or adults, or at 18°C. Exposure to chill coma tends to increase survival time during subsequent food deprivation in females (Figure 5C). Previous exposure to heat shock extends chill coma recovery time. Recovery is also delayed as a result of aging, growth at 28°C, previous exposure to starvation stress, growth on ethanol, menadione sodium bisulfite, or high sugar-supplemented medium, and maintenance at high density as adults (Figure 5D).
Whereas caloric restriction extends lifespan [19]–[20], a single 24 h food deprivation period does not affect lifespan. The increase in starvation stress resistance following chill coma recovery may be due to slowing of intermediary metabolism during chill coma and, consequently, preservation of metabolic energy.
Phenotypic Plasticity of Transcripts Correlated with Organismal Phenotypes
We used regression to identify Class I transcripts associated with variation in organismal phenotypes across the 20 environmental conditions, and MMC to construct environmentally correlated modules [13] of these transcripts (Figure 6). Phenotypic plasticity in development time is associated with 426 transcripts, of which 411 cluster into 36 highly correlated (average |r|>0.5) modules (Figure 6A). Similarly, phenotypic plasticity in lifespan, starvation stress resistance and chill coma recovery is associated with, respectively, 186, 320 and 440 transcripts, which cluster into 12, 30 and 23 highly correlated (average |r|>0.5) modules, respectively (Figure 6B–6D, Tables S4, S5, S6, S7). Modules associated with each trait show high degrees of inter-correlation, and there is evidence for cross-module clustering, indicating hierarchical co-regulation of the Class I plastic genes (Figure 6). We found little overlap (∼3%) between transcripts associated with genetic variation in lifespan, starvation resistance, and chill coma recovery under the standard growth condition [12] and transcripts associated with phenotypic plasticity of these traits. Since 1,125 of the 1,133 (99.3%) Class I transcripts are also genetically variable, the lack of concordance between the association with genetic and environmental phenotypic variation cannot be attributed to the trivial explanation that the genetically variable and phenotypically plastic transcripts do not overlap.
![Partitioning of correlated Class I phenotypically plastic transcripts associated with organismal phenotypes across different rearing conditions by MMC <em class="ref">[13]</em>.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/f16368d9a4c2f86f51f76263b3de0318.png)
Some modules associated with different organismal phenotypes are enriched for common transcripts, indicating pleiotropy for phenotypic plasticity (Figure S3). For example, the chill coma recovery module 17 contains the same transcripts as modules associated with phenotypic plasticity in development time and in starvation stress resistance. Whereas pleiotropy at the level of covariant transcriptional modules is prominent between chill coma recovery, starvation stress resistance and development time, it is sparser between lifespan and the other traits (Figure S3).
In summary, clustering analysis of Class I transcripts reveals a fragmented modular organization and distinct environmentally-responsive transcriptional signatures for the four fitness-related traits.
Class II Phenotypically Plastic Transcripts
To assess the relationship between genetic variation and phenotypic plasticity, we compared the previously reported genetic variance and micro-environmental variation (within-line variation) across the 40 inbred lines [12] from which our outbred population was derived, reared under the standard growth condition, with the variation in phenotypic plasticity (macro-environmental variation) and within-treatment variation of the same transcripts in the outbred population. We quantified genetic variation as the coefficient of variation among lines (CVL) and variation in phenotypic plasticity as the coefficient of macroenvironmental variance (CVME). We found a strong correlation between genetic variation and variation in phenotypic plasticity for Class I transcripts in both sexes (Figure 7A, 7B).

This comparison revealed an additional group of 982 environmentally sensitive transcripts with high macroenvironmental variation, but low genetic variance (Figure 7A, 7B, Figure S4, Table S1). These phenotypically plastic transcripts, which we designate as Class II, were not identified by our initial analysis due to high within-treatment environmental variation (quantified as the coefficient of variation within environments, CVEW, Figure S5A–S5D). Phenotypic plasticity for Class II transcripts was mostly sexually dimorphic, with 230 transcripts specific to females, 560 specific to males, and 192 in common for both sexes (Table S1). Class I transcripts have greater genetic variation for both sexes than environmentally robust transcripts, which are relatively stably expressed both across genotypes and treatments (Figure 7G, 7H, Figure S4C, S4D). In males the average genetic variance of Class II transcripts is lower than both Class I and robust transcripts (Figure 7G, Figure S4A), while in females the average genetic variance of Class II transcripts is lower than the Class I but higher than the robust transcripts (Figure 7H, Figure S4B). In contrast to the genetic variance, the average macroenvironmental variation across treatments of Class II transcripts is ∼two-fold greater than that of Class I transcripts for both sexes (Figure 7G, 7H, Figure S4C, S4D ).
There is little correlation between the level of genetic (Figure 7C, 7D) and macroenvironmental (Figure 7E, 7F) variation with the mean level of gene expression across all environments for Class I and robust transcripts. However, the macroenvironmental variance (Figure 7E, 7F) as well as the variance in gene expression within treatments (Figure S5A–S5D) and within inbred lines (Figure S5E, S5F) are correlated with the mean expression across treatments. To exclude the possibility that this observation is an artifact due to array quality, we examined the correlation between the previously published mean expression levels of transcripts across the 40 inbred lines [12] and the mean expression level in the outbred population across conditions. Transcript means were highly correlated between the two experiments (r = 0.960 and r = 0.963, for females and males, respectively; Figure S5G, S5H).
Since Class II transcripts exhibited sexual dimorphism in phenotypic plasticity, we evaluated the relationship between sexual dimorphism in mean gene expression across all 20 environments, and sexual dimorphism for phenotypic plasticity, for Class I and Class II transcripts as well as a sample of robust transcripts (Figure S6). We found a clear inverse relationship between sexual dimorphism for mean expression and sexual dimorphism for the variance in expression across environments for Class I transcripts, but not the other categories. Female-biased Class II genes for mean expression are male-biased for plasticity in expression, and vice versa.
Class II phenotypically plastic transcripts can be further classified into high and low expression categories. Highly expressed transcripts in females overlap transcripts with low expression in males, and GO analysis shows that these 19 transcripts encode yolk proteins and chorion proteins and are enriched for oogenesis and sexual reproduction (Table S8). Similarly, transcripts with low expression in females overlap transcripts with high expression in males, and GO analysis shows that these 162 transcripts encode male-specific proteins, accessory gland proteins and hormones which affect mating and post-mating behaviors (Table S9). Further GO analyses indicate that female-specific Class II transcripts are enriched in mitochondria - and muscle-related functions, whereas male-specific transcripts are enriched in functions of cuticular structure and DNA replication in meiosis (Tables S10 and S11). Enrichment of Class II transcripts for reproductive functions suggests that the high environmental responsiveness of these transcripts may protect reproductive fitness.
Conservation of Phenotypically Plastic Genes
To assess to what extent phenotypically plastic genes are evolutionarily conserved compared to the rest of the genome, we looked at the percentage of homologues across 12 Drosophila species, the ratio of non-synonymous to synonymous substitutions (ω) and fraction of adaptive fixations (α) using D. yakuba as outgroup [21]–[23] (Figure 8, Figure S7). Class I genes are less conserved across the Drosophila clade and have less constraints under selection than the environmentally robust genome. Previously, we documented plasticity of the Drosophila chemoreceptor repertoire [24]. Like chemoreceptor genes, many of the Class I transcripts also belong to rapidly evolving multigene families (Figure 8). Such rapid evolution may involve gene duplication and subfunctionalization, as is evident within the Cyp gene family [25]–[29].
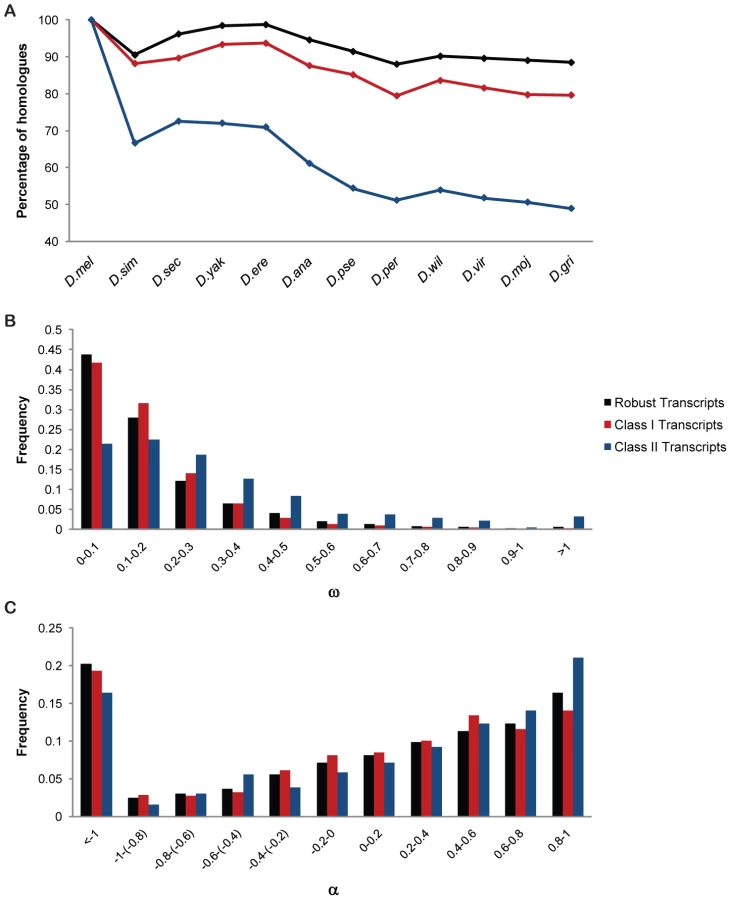
Class II genes show an even faster rate of evolution compared to robust transcripts with a significantly higher proportion of positively selected sites, as evident from the distributions of ω and α (Figure 8). Thus, these phenotypically plastic genes appear especially responsive to natural selection.
Discussion
Genome-wide transcriptional analysis of flies reared under 20 environmental conditions shows that ∼15% of the transcriptome exhibits phenotypic plasticity, while the rest is environmentally canalized. Logistical and economic constraints have limited this initial investigation to whole flies. We surveyed the FlyAtlas database [30] and found that transcripts associated with all organismal phenotypes are generally expressed in multiple, but not all tissues (Figure S8). In future studies it would be of interest to examine directly tissue-specific environmental modulation of expression of phenotypically plastic transcripts. Since we only examined adult flies, we could not detect transcripts that may show environmental plasticity at different developmental stages. Furthermore, although we provided a comprehensive analysis of the transcriptional response to a vast variety of conditions and treatments, additional treatments, e.g. different chemical exposures or sleep deprivation, might reveal additional features of the phenotypically plastic transcriptome. However, results from previous studies on genome-wide transcriptional responses to environmental and physiological changes in Drosophila are in line with our observations [6], [8], [11]. A previous study that examined phenotypic plasticity of the transcriptome during aging and upon exposure to paraquat-induced oxidative stress reported altered regulation of Hsp26, several metabolic enzymes, and glutathione-S-transferases [6]. Genome-wide transcriptional profiling during aging under conditions of caloric restriction also showed plastic responses of genes associated with xenobiotic defense and reproduction [8]. In addition, phenotypically plastic transcripts associated with xenobiotic defense, metabolism and chitin biosynthesis have been identified in Drosophila populations from tropical and temperate zones in Eastern Australia in response to temperature [11]. The analysis presented here is the most comprehensive study of phenotypic plasticity to date, which capitalizes on the unique properties of an outbred population reconstructed from well characterized inbred wild-derived lines, which enabled us to discriminate two classes of plastic transcripts.
Class I transcripts are not only phenotypically plastic, but are more genetically variable and evolve more rapidly than the rest of the transcriptome. Class I transcripts are enriched in functions of detoxification, metabolism, proteolysis and heat shock proteins. Class I transcripts also encode gene products of unknown function, including non-coding RNAs, which may contribute to modulation of chromatin structure and transcriptional regulation. The coupling of high genetic variation within a population and rapid evolution suggests interesting evolutionary forces acting on these genes.
Class II transcripts have low genetic variance for mean expression levels, but greater environmental variation in transcript abundance, and are even more rapidly evolving than Class I transcripts. It is tempting to speculate that reduced genetic variation for these transcripts within a population is the consequence of selection favoring genotypes with high phenotypic plasticity within each species, but with variable selection pressures across species [31]. Under this hypothesis genotypes with high transcriptional plasticity would be fixed within a species but divergent across species, which implies there is genetic variation in phenotypic plasticity on which selection acts. We note, however, that non-additive effects may confound inferences based on comparing an outbred population in many environments with inbred genotypes in one environment.
Two models of the genetic basis of phenotypic plasticity have been postulated [32]. Under the ‘allelic sensitivity’ model, the same alleles affect the mean value of a phenotype and its plastic response to environmental variation. Under the ‘gene regulation’ model, plasticity is a trait in itself, under the control of regulatory loci which modulate the expression of other genes in different environments. Our comparison of transcripts for phenotypic plasticity in an outbred population with genetically variable transcripts among the 40 inbred genotypes from which the outbred population was derived support the gene regulation hypothesis. We found no more overlap than expected by chance between transcripts associated with the mean and plasticity of four fitness-related traits. Class II phenotypically plastic transcripts are highly sexually dimorphic, but male-biased plastic transcripts are associated with female-biased mean expression levels, and vice versa, again suggesting an uncoupling between the mean and macroenvironmental variance. The Class I plastic transcripts cluster into modules of highly correlated transcripts, with a high degree of correlation across modules, further implicating co-regulation of plastic responses to environmental variation. Whereas this initial comprehensive survey of phenotypic plasticity is necessarily largely descriptive, it provides a foundation for future studies aimed at testing mechanisms that link environmental inputs to alterations in gene expression. It is likely that the environmentally plastic transcriptional regulators which we identified (Figure 4) will play a role in mediating these responses. Furthermore, since we did not consider the effects of DNA sequence variants on phenotypic plasticity, co-regulated modules of phenotypically plastic transcripts are undirected. Deriving causal transcriptional networks for genetic variation in phenotypic plasticity requires superimposing genetic variation [33]. The recent availability of whole genome sequences of the wild-derived inbred lines from the Raleigh population will enable such analyses in the future [23].
Materials and Methods
Fly Rearing
We generated a synthetic outbred population by round-robin crossing of 40 wild derived inbred lines of the Drosophila Genetic Reference Panel (DGRP) [12], [23], followed by random mating for over 47 generations. For age-synchronization, we randomly collected 1000 females and 1000 males and allowed oviposition for 12 h on grape agar plates supplemented with yeast paste. Unless indicated otherwise, 55 eggs were collected and subjected to different treatments throughout development. The standard rearing condition (cornmeal (65 g/L) -molasses (45 ml/L) -yeast (13 g/L) - agar medium at 25°C, 60–75% relative humidity and a 12 h light-dark cycle) resulted in hatching of ∼50 larvae. Adults were collected immediately after eclosion, and placed at a density of 25 females and 25 males under the desired condition. Flies were transferred onto fresh medium every two days.
Experimental Treatments
For nutritional and pharmacological treatments, flies were reared on standard medium supplemented with 225 ml/L molasses (‘high sugar’), 65 g/L yeast (‘high yeast’), 225 ml/L molasses and 65 g/L yeast (‘high sugar-high yeast’), 10% (v/v) ethanol, 200 µM fluoxetine hydrochloride, 47 mM dopamine, 1 mM nicotine, 2 mM caffeine or 4 mM menadione sodium bisulfite. Different physical environments included constant light, 28°C (‘high temperature’), 18°C (‘low temperature’), and exposure to different stresses, including heat shock (37°C for 1 h; 1 h recovery prior to RNA extraction), chill coma (3 h on ice; 1 h recovery prior to RNA extraction), and 24 h starvation. Different social environments included larval crowding (300 eggs/vial) and adult crowding (80 females and 80 males were pooled in each vial immediately after eclosion). To compare mated with non-mated flies, 50 single sex virgins were reared separately. Flies reared under standard conditions were mated. Aged flies were 30 days old.
Whole-Genome Transcript Analysis
We used Affymetrix Drosophila 2.0 arrays to assess whole genome transcriptional profiles. Males and females (3–5 days old) were collected between 1 : 00–3 : 00 pm by aspiration and immediately frozen on dry ice. RNA was extracted from three independent samples (30 flies/sex/condition), and 10 µg of biotinylated, fragmented cRNA was hybridized to each microarray. RNA extraction, labeling and hybridization were randomized across samples. Raw data were log2 transformed and normalized across sexes and conditions using a median standardization. For each probe set, we used the median log2 signal intensity as the measurement of expression. We used negative control probe sets to estimate background intensity. Probe sets with hybridization intensities below background under all different treatment conditions were removed from the analysis. We did not correct for probe mismatches due to segregating polymorphisms in the reconstituted outbred population, because (1) the average hybridization bias will be identical across all environmental conditions, and; (2) only about 3,000 single feature polymorphisms (SFPs) were identified among the original 40 inbred lines previously and their removal from the data set did not significantly influence the hybridization results [12]. Microarray data have been deposited in the ArrayExpress database (accession: E-MTAB-639 and are also available on the DGRP website (http://dgrp.gnets.ncsu.edu/).
We analyzed array data using a Generalized Linear Model (GLM) in SAS to partition phenotypic variation between sexes (S, fixed), environments/treatments (E, fixed), the S×E interaction (fixed) and the error variance (ε). To identify environmentally responsive Class I transcripts we used an FDR<0.05 to correct for multiple tests. Post-hoc LSD tests were used to identify transcripts with a significant environment term. Signal intensities for those transcripts were sex-centered by subtracting the female or male mean across all conditions for each gene. Transcripts with a significant interaction term were excluded.
To resolve Class II transcripts, we applied two filters. First, we selected transcripts with cross-treatment (macroenvironmental) variance >95th percentile of the macoenvironmental variance distribution of the Class I transcripts (coefficients of variation across treatments >7.06 and >7.12 for females and males, respectively). We filtered these transcripts further using an FDR>0.0001 for genetic variation among DGRP lines [12] for females and males separately. Finally, we removed overlapping transcripts between Class I and Class II. We used a form of K-means clustering (K = 2) to partition the Class II transcripts into groups of high and low expression. Specifically, for each sex we exhaustively identified the unique bipartition of Class II transcripts that minimized the total within group sum-of-squares.
The Modulated Modularity Clustering (MMC) algorithm [13] was used to group transcripts into covariant modules. Gene annotations were based on Flybase, version 5.36. Gene ontology analysis was done using the DAVID bioinformatics database, using the Benjamini correction of p<0.05 as criterion for enrichment [34], [35].
Organismal Phenotypes
To measure development time, we allowed flies to lay eggs for 3 hours (10 : 00am–1 : 00pm), after which 55 eggs were collected and placed under 14 different growth conditions (300 eggs were collected to assess development time under the larval crowding condition). We counted flies, sexes separately, that eclosed every 12 hours (N = 4 vials/condition). Life span was measured by collecting three females and three males immediately 1–3 days after eclosion, transferring them to fresh vials every 2–3 days, and recording survival daily (N = 26 replicates/condition). To measure starvation stress resistance, we placed ten 3–5 days old flies in vials containing 1.5% agar, and scored survival every 8 hours (N = 4×10/sex/condition). To measure chill coma recovery, we placed 3–7 day-old flies in empty vials on ice for 3 hours, and determined their subsequent recovery time at room temperature by their ability to recover upright posture (N = 2×50 flies/sex/condition). Phenotypic data are available on the DGRP website (http://dgrp.gnets.ncsu.edu/).
Transcript-Phenotype Associations
We used regression to identify transcripts with variation in expression levels that associated with organismal phenotypic variation (p<0.05). For traits with a significant sex by environment interaction, regression was applied to sexes separately (Y = μ+Exp+ε, where Exp denotes the covariate median log2 expression level). For traits without a significant sex by environment interaction, we used sex-centered measures of deviations from female or male means for both expression and organismal phenotypes. We used the residuals from the regressions (Y = μ+T+ε, where T denotes the trait covariate) to compute environmental correlations between transcripts significantly associated with each organismal phenotype for construction of covariant modules using MMC [13].
Supporting Information
Zdroje
1. BradshawAD 1965 Evolutionary significance of phenotypic plasticity in plants. Advances in Genetics 13 115 155
2. WaddingtonCH 1942 Canalization of developmnet and the inheritance of acquired characters. Nature 150 563 565
3. WaddingtonCH 1959 Canalization of development and genetic assimilation of acquired characters. Nature 183 1654 1655
4. GibsonG 2009 Decanalization and the origin of complex disease. Nat Rev Genet 10 134 140
5. LeeCKWeindruchRProllaTA 2000 Gene-expression profile of the ageing brain in mice. Nat Genet 25 294 297
6. ZouSMeadowsSSharpLJanLYJanYN 2000 Genome-wide study of aging and oxidative stress response in Drosophila melanogaster. Proc Natl Acad Sci U S A 97 13726 13731
7. CaoSXDhahbiJMMotePLSpindlerSR 2001 Genomic profiling of short - and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci U S A 98 10630 10635
8. PletcherSDMacdonaldSJMarguerieRCertaUStearnsSC 2002 Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol 12 712 723
9. SambandanDCarboneMAAnholtRRHMackayTFC 2008 Phenotypic plasticity and genotype by environment interaction for olfactory behavior in Drosophila melanogaster. Genetics 179 1079 1088
10. VinuelaASnoekLBRiksenJAKammengaJE 2010 Genome-wide gene expression regulation as a function of genotype and age in C. elegans. Genome Res 20 929 937
11. LevineMTEckertMLBegunDJ 2011 Whole-genome expression plasticity across tropical and temperate Drosophila melanogaster populations from Eastern Australia. Mol Biol Evol 28 249 256
12. AyrolesJFCarboneMAStoneEAJordanKWLymanRF 2009 Systems genetics of complex traits in Drosophila melanogaster. Nat Genet 41 299 307
13. StoneEAAyrolesJF 2009 Modulated modularity clustering as an exploratory tool for functional genomic inference. PLoS Genet 5 e1000479 doi:10.1371/journal.pgen.1000479
14. BushatiNCohenSM 2007 microRNA functions. Annu Rev Cell Dev Biol 23 175 205
15. RoySErnstJKharchenkoPVKheradpourPNegreN 2010 Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330 1787 1797
16. SambucettiPLoeschckeVNorryFM 2006 Developmental time and size-related traits in Drosophila buzzatii along an altitudinal gradient from Argentina. Hereditas 143 77 83
17. MenschJLavagninoNCarreiraVPMassaldiAHassonE 2008 Identifying candidate genes affecting developmental time in Drosophila melanogaster: pervasive pleiotropy and gene-by-environment interaction. BMC Dev Biol 8 78
18. ChapmanTLiddleLFKalbJMWolfnerMFPartridgeL 1995 Cost of mating in Drosophila melanogaster females is mediated by male accessory gland products. Nature 373 241 244
19. SohalRSWeindruchR 1996 Oxidative stress, caloric restriction, and aging. Science 273 59 63
20. FontanaLPartridgeLLongoVD 2010 Extending healthy life span–from yeast to humans. Science 328 321 326
21. ClarkAGEisenMBSmithDRBergmanCMOliverB 2007 Evolution of genes and genomes on the Drosophila phylogeny. Nature 450 203 218
22. LarracuenteAMSacktonTBGreenbergAJWongASinghND 2008 Evolution of protein-coding genes in Drosophila. Trends Genet 24 114 123
23. MackayTFCRichardsSStoneEABarbadillaAAyrolesJF 2012 The Drosophila melanogaster Genetic Reference Panel. Nature in press
24. ZhouSStoneEAMackayTFCAnholtRRH 2009 Plasticity of the chemoreceptor repertoire in Drosophila melanogaster. PLoS Genet 5 e1000681 doi:10.1371/journal.pgen.1000681
25. StrodeCWondjiCSDavidJPHawkesNJLumjuanN 2008 Genomic analysis of detoxification genes in the mosquito Aedes aegypti. Insect Biochem Mol Biol 38 113 123
26. FeyereisenR 2006 Evolution of insect P450. Biochem Soc Trans 34 1252 1255
27. ClaudianosCRansonHJohnsonRMBiswasSSchulerMA 2006 A deficit of detoxification enzymes: pesticide sensitivity and environmental response in the honeybee. Insect Mol Biol 15 615 636
28. RichardsSGibbsRAWeinstockGMBrownSJDenellR 2008 The genome of the model beetle and pest Tribolium castaneum. Nature 452 949 955
29. RansonHClaudianosCOrtelliFAbgrallCHemingwayJ 2002 Evolution of supergene families associated with insecticide resistance. Science 298 179 181
30. ChintapalliVRWangJDowJA 2007 Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39 715 720
31. ZhangXSHillWG 2007 Competition can maintain genetic but not environmental variance in the presence of stabilizing selection. Evolution 61 1532 1545
32. ViaSGomulkiewiczRDe JongGScheinerSMSchlichtingCD 1995 Adaptive phenotypic plasticity: consensus and controversy. Trends Ecol Evol 10 212 217
33. MackayTFCStoneEAAyrolesJF 2009 The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10 565 577
34. Huang daWShermanBTLempickiRA 2009 Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 44 57
35. Huang daWShermanBTLempickiRA 2009 Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37 1 13
36. YuJPacificoSLiuGFinleyRLJr 2008 DroID: the Drosophila Interactions Database, a comprehensive resource for annotated gene and protein interactions. BMC Genomics 9 461
37. MuraliTPacificoSYuJGuestSRobertsGG3rd 2011 DroID 2011: a comprehensive, integrated resource for protein, transcription factor, RNA and gene interactions for Drosophila. Nucleic Acids Res 39 D736 743
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2012 Číslo 3
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database