
Lymfangiomatóza – vzácné generalizované onemocnění lymfatického systému
Lymphangiomatosis – very rare disease of the lymphatic vessels
Lymphangiomatosis is rare disease, we can find this entity in differential diagnosis of osteolytic leasions of bones of unknown origin. Typical sign for lymphangiomatosis is proliferation of lymphatic tissue with production of lymphangiomas in various organs and systems. Clinical manifestation of disease is variable, involvement of lungs and bone is typical. In our article we present recent classification of lymphatic tissue neoplasias, their clinical symptoms and treatment possibilities.
Keywords:
lymphangiomatosis
Autori:
Zdeněk Král 1; Marta Krejčí 1; Ivana Červinková 2; Luděk Pour 1; Martin Štork; Martin Krejčí 1; Viera Sandecká 1; Zdeněk Adam 1
Pôsobisko autorov:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Klinika dětské radiologie LF MU a FN Brno
2
Vyšlo v časopise:
Vnitř Lék 2021; 67(E-4): 9-12
Kategória:
Súhrn
Lymfangiomatóza je vzácné onemocnění, se kterým se můžeme setkat při diferenciální diagnostice osteolytických změn skeletu nejasné etiologie. Je způsobené proliferací tkáně lymfatických cest s tvorbou lymfangiomů. Klinická manifestace je různorodá, časté bývá postižení plic a kostí, ale i dalších tkání. Následující text je věnován recentní klasifikaci neoplazií z lymfatických cest, jejich klinickým projevům a léčebným možnostem.
Klíčová slova:
lymfangiomatóza
Úvod
Lokalizované bujení lymfatických cest, takzvaný lymfangiom, není úplně vzácné onemocnění. Při pohledu do databáze české a slovenské literatury s názvem „medvik.cz“ se po zadání termínu „lymfangiom“ v říjnu 2020 objevilo 90 záznamů. Bujení lymfatických cest přesahující jeden orgán se nazývá lymfangiomatóza, tedy podobný termín jako hemangiomatóza, jenže v tomto případě jde o proliferaci buněk lymfatických cév. Při zadání termínu „lymfangiomatóza“ do databáze české a slovenské literatury jsme našli pouze 4 publikace, které se tomuto tématu věnují (1–4). A proto považujeme za vhodné tuto vzácnou jednotku následujícím textem připomenout, přiblížit současnou klasifikaci neoplazií z lymfatických cest, klinické projevy a také léčebné možnosti. Problém generalizované hemangiomatózy jsme ve Vnitřním lékařství již popsali, s generalizovanou lymfagniomatózou jsme se na našem pracovišti setkali 1× za 30 let, a proto považujeme za vhodné na tuto jednotku upozornit, protože kvalitu života těchto nemocných zásadně zlepší časná diagnostika a léčba. Tuto jednotku nutno zvažovat vždy při diferenciální diagnóze nejasné osteolýzy.
Klasifikace neoplazií z lymfatických cest a klinické projevy
Dříve se pro lokalizované bujení lymfatických cest používal termín lymfangiom a pro bujení lymfatických cév, přesahující jeden orgán, pak termín lymfangiomatóza. Jenže klasifikace všeho se vyvíjejí od jednoduchých směrem ke stále složitějším, a tak je tomu i s cévními anomáliemi. Odborníky na cévní anomálie sdružuje celosvětová organizace zvaná International Society for the Study of Vascular Anomalies (ISSVA) a tato organizace postupně upravuje klasifikace cévních malformací (5–7). Podrobnosti lze najít na jejich internetové adrese https:issva.org. Tabulka 1 uvádí ISSVA klasifikaci lymfatických vaskulárních abnormalit z roku 2018.

Pro „chanel typ lymphatic malformation“ jsme nenašli akceptovaný český ekvivalent, tak v dalším textu ponecháme anglickou formu názvu. Podobně jako cévní malformace, tak se i lymfatické malformace manifestují převážně již v dětském věku, u dospělých pacientů se s nimi lékaři setkávají méně často. Mohou mít ale značně devastující důsledky pro svého nositele, jak popisují četní autoři (7–9).
V etiologii těchto malformací má důležité místo vaskulární endoteliální růstový faktor (VEGF), dále genetické abnormality MAPK signální cesty a mTOR signální cesty. Jejich zvýšená aktivita má etiopatogenetickou roli u těchto procesů, a proto je i cílem léčebných intervencí (10–14). Společnou charakteristikou těchto nemocí je alterace lymfatické tkáně, vznik osteolytických ložisek, poškození hrudního mízovodu (ductus thoracicus) a vznik výpotků (chylózní výpotek v pleurální či perikardiální dutině, případně v peritoneální dutině). Bujení lymfatických cév může být podkladem patologických mas v mediastinu, případně i v retroperitoneu. Ke kožním projevům pak patří vznik lymfedému. Pacienti s komplexní lymfatickou anomálií mohou mít hypoalbuminemii, hypoproteinemii, hypogamaglobulinemii a také lymfocytopenii. Někdy tyto abnormality provází také trombocytopenie, nízká koncentrace fibrinogenu a zvýšená hodnota D-dimerů, tedy známky chronické lokalizované intravaskulární koagulopatie. Příčina koagulopatie zde není příliš jasná. Charakteristiku těchto klinických jednotek, dříve zahrnovaných pod termín „lymfangiomatóza“, které se ale navzájem překrývají, ilustruje tabulka 2.

V případě nemoci Gorhama – Stauta (syndrom GS) je třeba vyloučit jiné příčiny osteolýzy. U pacientů se syndromem GS je typická vysoká hodnota kostní formy alkalické fosfatázy a sérová hladina CTX1. Aplikace bisfosfonátů vede k poklesu těchto markerů (15, 16).
Počet postižených kostí bývá vyšší u lymfangiomatóz zařazených pod termín generalizovaná lymfatická anomálie a kaposiformní lymfangiomatóza, než v případně syndromu GS. Osteolýza v případě nemoci GS je obvykle lokalizovaná v lebce, žebrech, klíčku, či v krční páteři. Osteolytická ložiska v případě GS syndromu mají progresivní charakter, zatímco osteolytická ložiska při generalizované lymfatické anomálii či kaposiformní lymfangiomatóze tak jasnou tendenci k rozšiřování a zvětšování nemají. Pacienti s generalizovanou lymfatickou anomálií a kaposiformní lymfangiomatózou mají častěji postiženu bederní páteř než pacienti s nemocí GS (17).
Pro generalizovanou lymfatickou anomálii jsou typickými příznaky pleurální a perikardiální výpotky a mediastinální masy. Výpotek přitom může být jak hemorrhagický, tak i chylózní. Progresivní zhoršování celkového stavu, přítomné retroperitoneální masy a hemorrhagické výpotky spojené s trombocytopenií provázejí často kaposiformní lymfangiomatózou (7–9). Trombocytopenie a koagulační poruchy se vyskytují dominantně u kaposiformní angiomatózy, která má nejvíce agresivní průběh z popsaných poruch (7–9).
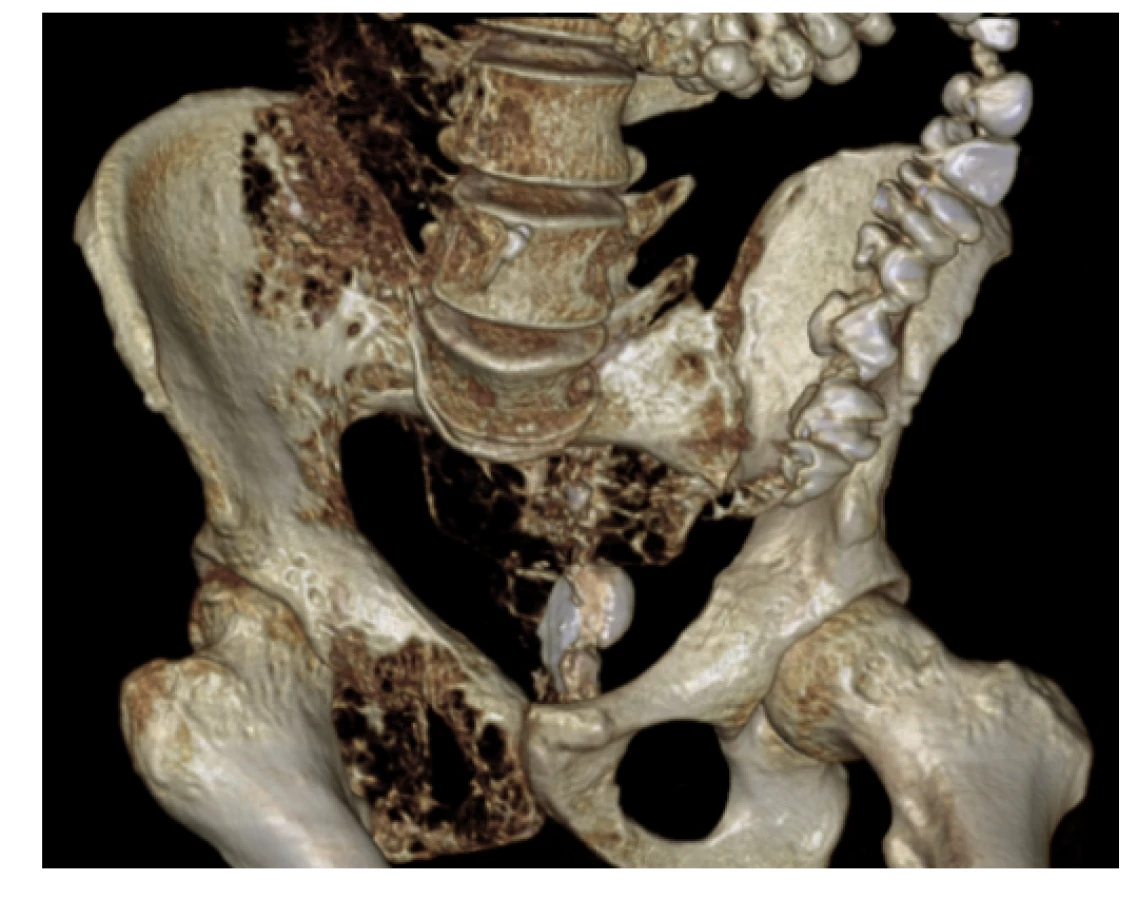
Na našem pracovišti jsme se s generalizovanou lymfangiomatózou setkali pouze jednou. Šlo o mladého 27letého muže, který byl hospitalizován pro intenzivní bolesti v oblasti pánve a páteře. Tyto bolesti mu znemožňovaly chůzi. Krevní obraz měl v normě i základní koagulační vyšetření bylo v normě, bez známek hyperkoagulace. V dokumentaci z jiného pracoviště, kterou měl u sebe, byla histologicky ověřená diagnóza „lymfangiomatózy“. Tento pacient měl i přes adekvátní vysvětlení opakovaně negativní postoj k nabízeným diagnostickým a léčebným postupům, další diagnostiku a terapii odmítl a podepsal negativní reverz. Devastující následky jeho dlouholeté choroby ilustrují obrázky 1–7.






Léčba
Všechny dále zmíněné terapeutické alternativy jsou jen částečně účinné. Nejdůležitější léčbou je léčba podpůrná. V případě osteolýzy se považují za indikované bisfosfonáty. Byly testovány samostatně anebo v kombinaci s interferonem alfa a tato léčba měla prokazatelné zmírnění obtíží (18). Dále u pacientů s lymfangiomatózou byl použit, podobně jako u hemangiomatózy, bevacizumab, dále také propranolol a případně steroidy. Tyto léky mají prokázaný potenciál mírnit progresi nemoci, a tím i její symptomy, ale jejich efekt je limitovaný (19–21). V posledních letech se objevila role PIK3/AKT/mTOR signální cesty. A proto se začal těmto pacientům podávat mTOR inhibitor sirolimus, synonymem rapamycin, který inhibuje lymfangiogenezu a také metabolické procesy v lymfatických endoteliálních cévách (22–24). Sirolimus byl dokonce testován s výborným výsledkem v prospektivní klinické studii. Na léčbu pozitivně odpovědělo 7 ze 7 pacientů s generalizovanou lymfatickou anomálií, 3 z 3 pacientů s nemocí GS a 5 ze 7 pacientů s kaposiformní lymfangiomatózou (25). Sirolimus je dle pravidel Státního ústavu pro kontrolu léčiv (SÚKL) hrazen u pacientů po transplantaci ledvin jako profylaxe rejekce, pokud prokazatelně netolerují léčbu inhibitory kalcineurinu (včetně případů kalcineurinovými inhibitory vyvolané nefrotoxicity) nebo u pacientů s refrakterní rejekcí po orgánové transplantaci. Ale také pacientům s lymfangioleiomyomatózou (LAM) s plicním postižením. A tak je otázka, zda v případě lymfangiomatózy, což jistě je příbuzné onemocnění s lymfangioleiomyomatózou, je, či není nutno žádat revizní lékaře o schválení. Dávka cílové hladiny sirolimu není zcela jasně definována. Z jedné klinické studie vyplynula doporučená průměrná hladina 5–15 ng/ml (26). Pokud mediakamentózní léčba selhává, testuje se radioterapie a chirurgie. V případě chylothoraxu drenáže, případně ligace hrudního mízovodu (ductus thoracikus), nebo dokonce jeho embolizace (27).
Radioterapie je další alternativa. Má potenciál stabilizovat tuto nemoc a byla přínosná u 80 % pacientů. Použita byla dávka 40–45 Gy (28–29). Ale i menší dávky 16–20 Gy mohou pomoci při zvládání chylothoraxu či chyloperikardia (9). Podrobně zkušenosti s lokální léčbou rozvádějí němečtí autoři (30).
Závěr
Lymfangiomatóza může nabývat různých klinických projevů a může mít mutilující a devastující účinky na lidský organismus. Pro postižené nemocné bude stále nejdůležitější systematická podpůrná léčba. Vývoj v oblasti medikamentózní léčby, inhibující vaskulární bujení, bisfosfonáty a v tomto případě sirolimus neboli rapamycin mohou alespoň částečně zpomalit proces proliferace lymfatických cév. Zásadní pro úspěch léčby je častná diagnostika a časné zahájení léčby.
KORESPONDENČNÍ ADRESA AUTORA:
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika LF MU a FN Brno
Jihlavská 20, 625 00 Brno
Článek přijat redakcí: 2. 12. 2020
Článek přijat po recenzích k publikaci: 28. 5. 2021
Zdroje
1. Navrátilová Z, Štěrba J. Lymfangiomatóza skrota, břišní stěny a dolních končetin LYMPHO. Praha: AMCA. 2012: s. 26.
2. Lukeš J, Dort V, Kohoutek V. Kostní systémová lymfangiomatóza. Československá pediatrie. 1973; 28 : 417–419.
3. Krásný J Baráková D, Chodounský Z et al. Lymfangiom orbitopalpebrální oblasti. Česká a slovenská oftalmologie. 2014; 70 : 152–159.
4. Rygl M, Šnajdauf J, Pýcha K et al. Abdominální lymfangiomy v dětském věku. Rozhledy v chirurgii. 2000; 79 : 609–612.
5. International Society for the Study of Vascular Anomalies: ISSVA classification for vascular anomalies (approved at the May 2018 General Assembly in Amsterdam, the Netherlands). http://issva.org/classification (last accessed June 2018).
6. Trenor CC, Chaudry G. Complex lymphatic anomalies. Semin Pediatr Surg 2014; 23 : 186–190.
7. Blei F. Lymphangiomatosis: clinical overview. Lymphat Res Biol. 2011; 9 : 185–190.
8. Adams DM, Ricci KW. Vascular Anomalies: Diagnosis of Complicated Anomalies and New Medical Treatment Options. Hematol Oncol Clin North Am 2019; 33(3): 455–470.
9. Ozeki M, Fukao T. Generalized lymphatic anomaly and Gorham-Stout disease: overview and recent insights. Adv Wound Care (New Rochelle) 2019; 8 : 230–245.
10. Greene AK, Goss JA. Vascular anomalies: from a clinicohistologic to a genetic framework. Plast Reconstr Surg 2018; 141 : 709e–717e.
11. Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest 2014; 124 : 898–904.
12. Osborn AJ, Dickie P, Neilson DE et al. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Hum Mol Genet 2015; 24 : 926–938.
13. Boscolo E, Coma S, Luks VL et al. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2015; 18 : 151–162.
14. Manevitz-Mendelson E, Leichner GS, Barel O et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis 2018; 21 : 287–298.
15. Yerganyan VV, Body JJ, De Saint Aubain N, Gebhart M. Gorham-Stout disease of the proximal fibula treated with radiotherapy and zoledronic acid. J Bone Oncol 2015; 16 : 42–46.
16. Hammer F, Kenn W, Wesselmann U et al. Gorham-Stout disease – stabilization during bisphosphonate treatment. J Bone Miner Res. 2005; 20 : 350–353.
17. Kato H, Ozeki M, Fukao T, Matsuo M. MR imaging findings of vertebral involvement in Gorham-Stout disease, generalized lymphatic anomaly, and kaposiform lymphangiomatosis. Jpn J Radiol 2017; 35 : 606–612.
18. Kuriyama DK, McElligott SC, Glaser DW et al. Treatment of Gorham–Stout disease with zoledronic acid and interferon-alpha: a case report and literature review. J Pediatr Hematol Oncol 2010; 32 : 579–584.
19. Ozeki M, Funato M, Kanda K et al. Clinical improvement of diffuse lymphangiomatosis with pegylated interferon alfa-2b therapy: case report and review of the literature. Pediatr Hematol Oncol 2007; 24 : 513–524.
20. Grunewald TG, Damke L, Maschan M et al. First report of effective and feasible treatment of multifocal lymphangiomatosis (Gorham–Stout) with bevacizumab in a child. Ann Oncol 2010; 21 : 1733–1734.
21. Ozeki M, Fukao T, Kondo N. Propranolol for intractable diffuse lymphangiomatosis. N Engl J Med 2011; 364 : 1380–1382.
22. Hammill AM, Wentzel M, Gupta A et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer 2011; 57 : 1018–1024.
23. Lackner H, Karastaneva A, Schwinger W et al. Sirolimus for the treatment of children with various complicated vascular anomalies. Eur J Pediatr 2015; 174 : 1579–1584.
24. Reinglas J, Ramphal R, Bromwich M. The successful management of diffuse lymphangiomatosis using sirolimus: a case report. Laryngoscope 2011; 121 : 1851–1854.
25. Adams DM, Trenor CC 3rd, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016; 137 : 1–10.
26. Nadal M, Giraudeau B, Tavernier E et al. Efficacy and safety of mammalian target of rapamycin inhibitors in vascular anomalies: a systematic review. Acta Derm Venereo 2016; 96 : 448–452.
27. Itkin M. Interventional treatment of pulmonary lymphatic anomalies. Tech Vasc Interv Radiol 2016; 19 : 299–304.
28. Heyd R, Micke O, Surholt C et al. German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham–Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys 2011; 81 : 179–185.
29. Dunbar SF, Rosenberg A, Mankin H, Rosenthal D, Suit HD. Gorham's massive osteolysis: the role of radiation therapy and a review of the literature. Int J Radiat Oncol Biol Phys 1993; 26 : 491–497.
30. Müller-Wille R, Wildgruber M, Sadick M et al. Vascular Anomalies (Part II): Interventional therapy of peripheral vascular malformations. Rofo 2018; 190 : 927–937.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2021 Číslo E-4
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Perkutánní intervence chronického uzávěru koronární tepny – komu, kdy a proč
- Lymfangiomatóza – vzácné generalizované onemocnění lymfatického systému
- Aortálna stenóza a dysfunkcia pravej komory
- Pancytopenie jako hlavní projev nákazy lidským virem imunodeficience ve stadiu AIDS