
Selective Recruitment of Nuclear Factors to Productively Replicating Herpes Simplex Virus Genomes
HSV-1 is a human pathogen that infects over 50% of the population. The virus persists as a latent infection in the ganglia of an infected host and upon stressful conditions is reactivated to a lytic state in which it causes recurrent sores at the initial site of infection. During lytic infection, HSV highjacks the host cell to propagate its genome and produce new virus particles. However, there is limited knowledge of what cellular proteins interact with and function on the viral genome. We therefore developed methods to purify viral genomes from productively infected cells to identify associated viral and cellular proteins. We found proteins and protein complexes that have previously been implicated in HSV infection to be enriched on viral genomes, as well as several novel proteins that are likely involved in productive infection. These data provide valuable insight into HSV biology. Furthermore, these methods can be adapted to study other viruses, as well as other aspects of the HSV life cycle.
Published in the journal:
Selective Recruitment of Nuclear Factors to Productively Replicating Herpes Simplex Virus Genomes. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004939
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004939
Summary
HSV-1 is a human pathogen that infects over 50% of the population. The virus persists as a latent infection in the ganglia of an infected host and upon stressful conditions is reactivated to a lytic state in which it causes recurrent sores at the initial site of infection. During lytic infection, HSV highjacks the host cell to propagate its genome and produce new virus particles. However, there is limited knowledge of what cellular proteins interact with and function on the viral genome. We therefore developed methods to purify viral genomes from productively infected cells to identify associated viral and cellular proteins. We found proteins and protein complexes that have previously been implicated in HSV infection to be enriched on viral genomes, as well as several novel proteins that are likely involved in productive infection. These data provide valuable insight into HSV biology. Furthermore, these methods can be adapted to study other viruses, as well as other aspects of the HSV life cycle.
Introduction
The genomes of eukaryotic DNA viruses vary in complexity with respect to the number of genes they encode, and hence their dependence on host-cell functions. With the exception of poxviruses, all replicate in the cell nucleus and therefore utilize the nuclear machinery for the maintenance, replication, and expression of their genomes. The dynamic interactions between viral and cellular proteins and the viral genome, function to mediate the different steps in the life cycle of the virus, and hence determine the outcome of infection. These include interactions that mediate the entry of the genome into the nucleus, its expression and replication, and ultimately the packaging of nascent genomes in capsids.
Herpes simplex virus 1 (HSV-1) has a linear genome comprised of 152 kilobasepairs [1,2]. It enters the nucleus from the capsid through pores in the nuclear envelope [3–5]. The genome then participates in a series of interactions that results in a nucleo-protein complex near ND10 structures [6]. Here, the genome is susceptible to activities of the intrinsic cellular antiviral response. The genome also contains nicks and gaps, and these along with the genomic termini elicit a DNA damage response, the nature of which may be consequential to viral infection [7]. Viral genomes initially associate with ND10 structures, where through the action of ICP0, ND10 proteins are degraded or dispersed resulting in the prerequisite structure for efficient transcription and replication [6,8]. Viral DNA replication then results in the formation large replication compartments, which fill the host nucleus and concentrate viral and cellular factors to replicating viral genomes [9].
HSV-1 encodes two transcription factors, VP16 [10,11] and ICP4 [12], which function along with the cellular RNA polymerase II transcription machinery [13] to transcribe the viral genome. These factors initially colocalize with prereplicative genomes [14–16] and these interactions as well as those involving viral and cellular RNA-processing factors result in an ordered cascade of viral gene expression [17,18]. Seven HSV gene products are sufficient in cells to replicate DNA in an HSV-origin dependent manner [19]. While this set of viral proteins includes a DNA-dependent DNA polymerase and other functional analogs of cellular DNA replication proteins, it is not sufficient to drive origin-dependent replication in vitro, suggesting the requirement for as yet unknown cellular proteins [20]. Finally, an additional set of proteins interacts with the genome in the processes of cleaving unit length genomes and their packaging in capsids [21]. These processes have been, and continue to be the focus of studies in many laboratories since significant gaps exist in our understanding of all these processes, and how they ultimately contribute to viral multiplication and pathogenesis. A shortcoming contributing to these gaps is our relative lack of knowledge of the proteins, particularly cell-derived, which interact with viral genomes in different phases of infection.
Recently, ethynyl-modified nucleosides along with click chemistry and immunofluorescence were used to trace the fate of input adenovirus genomes in infected cells [22]. Nucleoside analogs were also incorporated into replicating herpes simplex and vaccinia viral DNA to demonstrate that this technique can be used to label other viral genomes and could potentially be used to track these genomes throughout infection. In addition, ethynyl-modified nucleosides have been used in a procedure known as isolation of proteins on nascent DNA (iPOND) to identify the proteins at cellular replication forks [23–28]. This procedure involves the metabolic incorporation of 5-ethynyl-2´-deoxyuridine (EdU) into the DNA, biotinylating the EdU-labeled DNA by click chemistry, followed by the affinity purification of the biotinylated DNA, and the subsequent analysis of the proteins associated with it. We have adopted and modified these procedures to enable the visualization of the HSV genome at different stages of infection, as well as the interrogation of the viral and cellular proteins on replicated/replicating viral genomes. The results elucidate the viral and cellular proteins associating with viral DNA during infection and provide a comprehensive view of the cellular machinery functioning on HSV genomes.
Results
Labeling and imaging HSV genomes
Ethynyl-modified nucleosides have been used to prelabel and then track single incoming adenovirus genomes within infected cells [22]. While this approach was also used to examine HSV genomes in replication compartments, input genomes were not imaged. We sought to determine if ethynyl-modified nucleosides could be used to label and track HSV genomes during early (before DNA replication), as well as late (after DNA replication) stages of infection. We also intended to use viral DNA imaging to optimize HSV genome labeling for purification of viral genomes by iPOND.
Preliminary experiments demonstrated that EdU and EdC were poorly incorporated in HSV DNA. We hypothesized that deletion of the HSV deoxyuridine triphosphatase (dUTPase) and uracil glycosylase enzymes would increase incorporation into the viral genome. HSV-1 uracil glycosylase and dUTPase mutant strains were generated by introducing premature termination codons early in the reading frames of the UL2 and UL50 genes (Fig A in S1 Text). As found for labeling of adenovirus genomes [22], ethynyl nucleoside incorporation into HSV genomes resulted in slightly reduced virus titers (Fig B in S1 Text). The same concentrations of EdU or EdC had a greater effect on the titer of the UL2/UL50 double mutant virus than on wild type KOS, suggesting that the double mutant is more efficiently labeled by both EdU and EdC. EdU was used in all subsequent experiments.
To compare the relative amount of EdU incorporated into wild type and mutant genomes, we carried out viral infection and DNA imaging as outlined in Fig 1A and 1B. EdU labeled input genomes and replication compartments were tagged with Alexa Fluor 488 by click chemistry and visualized by fluorescence microscopy (Fig 2). Prelabeled UL2/UL50 mutant genomes that colocalized with the viral transcription factor, ICP4, were visualized in the nucleus of infected cells (Fig 2A). The distribution of ICP4 foci two hours after infection largely resembles that observed previously after infection with HSV-1 at a MOI of 10 PFU/cell [29]. Using these same conditions, we were unable to detect KOS genomes prelabeled with 2.5 μM EdU (Fig 2A). We also visualized viral replication compartments that colocalize with ICP4 8 hpi in both wild type KOS and UL2/UL50 mutant virus infected cells (Fig 2B). While it was possible to detect EdU labeling coinciding with ICP4 staining in KOS infected cells, significantly more was observed with the UL2/UL50 mutant. Taken together, the UL2/UL50 mutant virus incorporates more EdU into its genome during DNA replication and allows for more sensitive imaging of HSV-1 viral DNA during infection.


Optimization and validation of HSV iPOND
To identify the viral and cellular proteins that function on viral DNA at different stages of infection, we adapted the iPOND method [25] for analysis of viral genomes and associated proteins. To optimize viral iPOND, we initially considered several factors. Proliferating cells grown in the presence of EdU incorporate EdU into their genomes during DNA replication (Fig 2B, uninfected, 2.5 μM). Therefore, conditions in which viral DNA, but not cellular DNA is labeled in the presence of EdU were established. Addition of EdU to the growth medium of proliferating Vero cells that were mock - or HSV-1-infected resulted in labeling of 65% or 29% of cellular genomes (Fig C in S1 Text, panels Vero cells), respectively. HSV infection inhibits G1/S and G2/M phases of the cell cycle [30,31] consistent with less labeling of cellular DNA in infected cells. In contrast to proliferating cells, less than 1% of cellular genomes were labeled with EdU when human MRC-5 fibroblast cells that were grown to confluency were infected with HSV in the presence of EdU (Fig C in S1 Text, panels MRC-5 cells). Therefore, resting MRC-5 cells were used in iPOND experiments to avoid contamination with cellular DNA. These cells also have the added advantage that they are a natural host to lytic HSV infection and they do not express high levels of cellular glycosylases to limit cleavage of labeled viral genomes [32].
One of the limitations of iPOND is that a large amount of EdU-labeled DNA is required to pull down enough protein for proteomic analysis [25]. Because the UL2/UL50 mutant virus is more efficiently labeled with EdU than wild type virus, we hypothesized that more viral DNA and associated proteins could be purified by iPOND of the mutant virus. We tested iPOND for the purification of proteins associated with genomes of wild type KOS, UL2 and UL50 single mutant, and UL2/UL50 double mutant viruses (outlined in Fig 1C). The relative protein yield for each virus was compared by western blot for the viral transcription factor ICP4 (Fig D in S1 Text). ICP4 associates with viral genomes throughout infection and is a good indicator of protein yield. The negative control was iPOND carried out on virus-infected cells incubated in the absence of EdU. For all viruses tested, ICP4 was not detected in the negative control, but was detected when iPOND was carried out on viral genomes that were labeled with EdU. The greatest relative amount of ICP4 was detected with the UL2/UL50 mutant virus, consistent with fluorescence imaging of labeled viral genomes (Fig 2B). Therefore, the UL2/UL50 mutant virus was used for iPOND experiments.
To identify the proteins associated with viral genomes by iPOND, we labeled viral DNA at three time points during DNA replication. EdU was added to the medium of infected cells at 4–6, 6–8, or 8–12 hpi and cells were fixed for iPOND at 6, 8, or 12 hpi, respectively. Proteins recovered by iPOND were probed for ICP4 by western blotting (Fig 3A). ICP4 was detected at all time points, but not in the unlabeled negative control. To ensure that DNA isolated by iPOND was viral, input DNA from cell lysates and DNA bound to streptavidin-coated beads was extracted, the amount of viral DNA was measured, and the percentage of viral/total DNA was calculated (Fig 3B). DNA eluted from beads during iPOND experiments was nearly 100% viral in nature. This is a significant enrichment compared to input DNA (0.2–1.5% viral). To determine if the entire viral genome was labeled and purified in our assays, high throughput DNA sequencing was carried out on DNA eluted from streptavidin-coated beads (Fig E in S1 Text). At all time points, the distribution of bead-bound DNA was relatively homogeneous across the viral genome. Taken together, iPOND should enable the specific purification of proteins associated with the entire replicated HSV-1 genome.

Factors that mediate host cell nuclear processes are enriched on HSV-1 genomes during viral DNA replication
To determine the identity of proteins bound to viral genomes at 6, 8, and 12 hpi, mass spectrometry was carried out on proteins that were crosslinked to viral DNA and purified by iPOND. Two independent iPOND experiments were carried out for each time point, each with an unlabeled virus negative control that was prepared on the same day with the same cells, virus, and reagents. Proteins were considered significantly enriched on viral genomes if they were identified with high confidence in duplicate experiments to be enriched by at least four fold over the negative control. The types of proteins identified at all three time points are summarized in Fig 3C and individual complexes and proteins are listed in Tables 1–6 and Table A in S1 Text. The most abundant types of proteins enriched on isolated viral DNA include those involved in RNA processing, transcription, chromatin remodeling, DNA repair, and DNA replication.
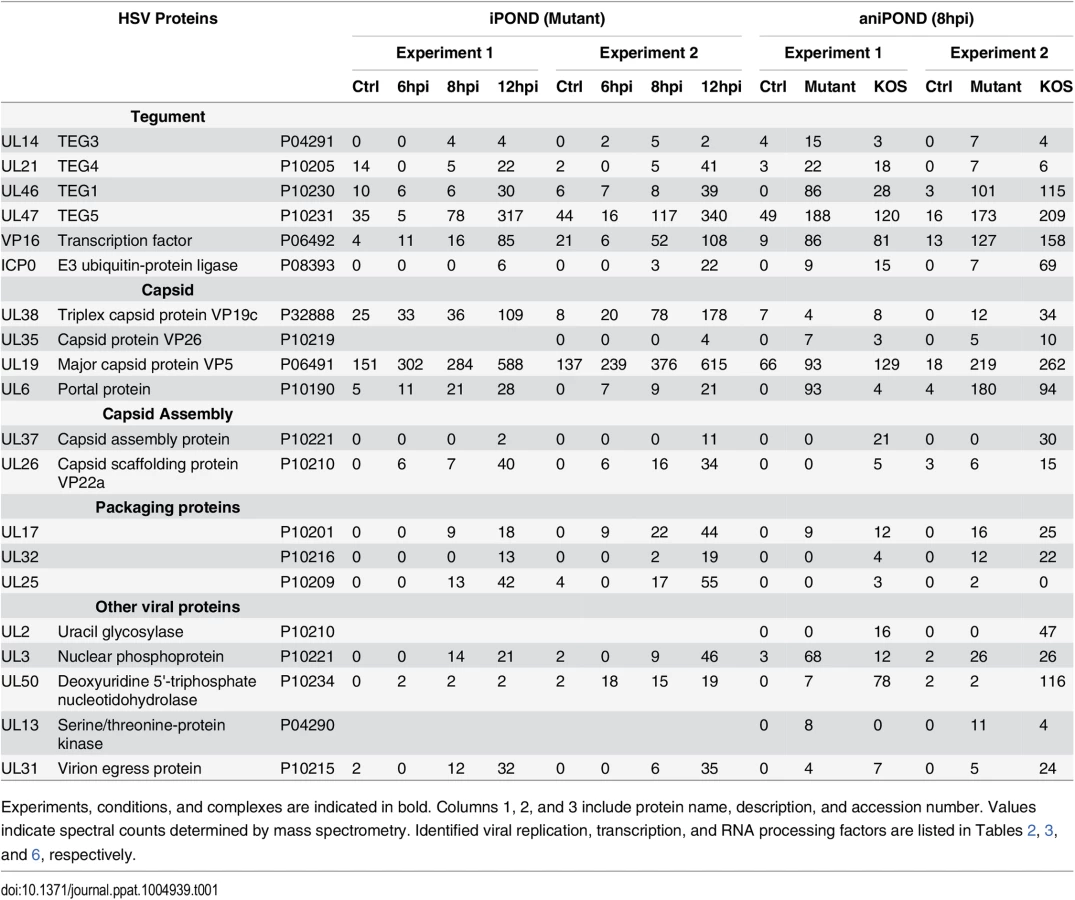





Furthermore, proteins that mediate nuclear transport, components of the nuclear cytoskeleton, and HSV structural proteins were bound to viral genomes. Several trends are present in these data. First, the total number of proteins that were recovered increased with time of infection. This is consistent with increasing amounts of labeled DNA as replication proceeds, allowing for more sensitive detection of bound proteins. Second, there was a relative increase in proteins that function in post-transcriptional RNA processing, as well as viral structural proteins with time. The increase in viral structural proteins including tegument proteins, capsid assembly factors, portal protein (UL6), and capsid proteins reflects the packaging of nascent genomes at later times during infection (Table 1).
Comparison of proteins identified at each time point suggests that the individual proteins found on replicated/replicating viral genomes at 6, 8, and 12 hpi were relatively similar (Fig 3D). There are significant overlaps between the three different time points with most proteins identified at two or more of the times sampled. The biggest difference was seen at 12 hpi and this reflects the increase in structural proteins, as well as the larger number of proteins recovered by iPOND at this time point.
Comparative analysis of replication proteins found on replicated cellular and viral DNA reveals the specificity of isolation of proteins on viral DNA (Table 2). Cellular replication forks are enriched for cellular replication factors including components of cellular DNA polymerase, clamp loader complex, MCM complex, as well as other replisome-associated proteins such as topoisomerases and PCNA [23,26,28]. In contrast, in our studies viral DNA was enriched for all seven components of the viral replication machinery including: ICP8, UL30 (polymerase), UL5/UL8/UL52 (helicase/primase complex), UL9 (origin binding protein), and UL42 (processivity factor). The cellular counterparts to these viral proteins were not enriched on viral genomes. One exception to this is the cellular processivity factor, PCNA. This protein was enriched on viral genomes at all times tested with the highest levels at 6 hpi, decreasing with time. Furthermore, cellular topoisomerases TOP1, TOP2a, and TOP2b were abundant on viral genomes and likely play a role in virus replication or other process.
aniPOND is an alternative method to purify viral genomes and associated proteins
Accelerated native iPOND (aniPOND) is a modified version of iPOND that is quicker and does not utilize crosslinking [33]. It involves native conditions during purification, while iPOND involves crosslinking and stringent wash conditions (Fig 1C and 1D). We therefore predicted that aniPOND would reveal a unique set of proteins involved in viral genome mechanics compared to iPOND because less direct interactors could be detected.
To obtain a more comprehensive view of proteins bound to viral genomes, we carried out aniPOND on KOS and UL2/UL50 mutant viruses that were incubated in the presence of EdU from 4–8 hpi and harvested at 8 hpi. Proteins eluted from viral DNA during aniPOND were assayed for ICP4 by western blotting (Fig 4A). ICP4 was detected when the infection was carried out in the presence of EdU (lanes 3 and 4), but not in the absence of EdU (lane 1). Importantly, using aniPOND it was also possible to recover ICP4 associated with wild type genomes, however, greater amounts where recovered in the sample with the mutant virus extract. In this experiment, significantly less (~2%) sample was required to isolate a similar amount of ICP4 to that recovered with iPOND. Therefore aniPOND is more efficient for the recovery of labeled viral DNA and associated proteins than iPOND. This is in agreement with comparison of the purification of replisome-associated proteins by each method [33].

To verify the specificity of aniPOND for the purification of replicated viral genomes, we carried out aniPOND on cells infected with the UL2/UL50 mutant virus that was maintained in the presence of acycloguanosine (ACG, acyclovir), a potent inhibitor of viral DNA replication. In the absence of viral DNA replication, DNA was not recovered by aniPOND and ICP4 was not detected by western blotting (Fig 4A, lane 2). To further validate aniPOND for purification of viral genomes, we determined the relative amount of viral DNA/total DNA purified by this method. DNA eluted from beads during aniPOND experiments was almost 100% viral in nature (Fig 4B, bound DNA, +EdU-ACG). This is a significant enrichment compared to the percent viral DNA present in lysates for this condition (input DNA, <2%). Very little DNA was detected when aniPOND was carried out on virus grown in the absence of EdU (bound DNA,-EdU) or in the presence of ACG (+ACG), consistent with specific purification of replicated viral DNA by aniPOND.
To determine the identity of proteins that copurified with viral genomes, mass spectrometry was carried out on samples prepared by aniPOND of labeled UL2/UL50 mutant and wild type KOS genomes at 8 hpi. Two independent experiments were carried out for each virus, with an unlabeled virus negative control that was done on the same day with the same cells and reagents. Almost twice as many proteins were identified with high confidence by aniPOND compared to iPOND at 8 hpi (184 compared to 96). The types of proteins identified by aniPOND are summarized in Fig 4C and individual proteins are listed in Tables 1–6 and Table A in S1 Text. Proteins that copurified with viral genomes by aniPOND at 8 hpi share the same functional categories as proteins that were purified by iPOND. In fact, pie charts that summarize the findings from these two experiments show very similar trends (compare Figs 4C to 3C 8hpi).
Proteins that copurified with UL2/UL50 mutant genomes by iPOND and aniPOND at 8 hpi were compared (Fig 4D). Fifty-three proteins were identified by both methods, 131 by only aniPOND, and 43 by only iPOND. Differences in proteins identified by each method likely reflect differences in the nature of DNA-protein interactions. For example, the viral helicase/primase complex was identified by iPOND but not aniPOND (Table 2). Crosslinking during iPOND could capture transient DNA-protein interactions or interactions that are lost during purification, which may be the case for ATPases such as the helicase/primase complex. On the other hand, the mediator of RNA polymerase II complex, as well as components of general transcription factor TFIID and TFIIH were identified by aniPOND but not iPOND (Table 3). Members of these complexes may not be in direct contact with the viral genome or may bind in an orientation that is not conducive to crosslinking. We have shown previously that the mediator complex, TFIID, and TFIIH copurify with ICP4 from virus-infected cells [34,35]. Here we also confirmed that ICP4 coprecipitates with mediator and TFIID from virus infected resting MRC-5 cells, along with a subset of transcription and chromatin remodeling factors that copurify with viral DNA (Table B in S1 Text). Therefore, ICP4 may provide a means to target these complexes to viral DNA.
Comparison of proteins identified by aniPOND of mutant genomes and wild type genomes (Tables 1–6, Mutant vs. KOS) reveal similar trends and in almost all cases the same proteins were found to be associated with both genomes. In fact, the most obvious difference is that viral peptides for UL2 and UL50 gene products were not enriched on UL2/UL50 mutant genomes but were enriched on wild type genomes (Table 1). This provides validation for these mutants not expressing UL2 and UL50 gene products and supports the use of mutant genomes for the purification and identification of virus-associated proteins.
To provide support for the specificity of iPOND and aniPOND methods for the purification of bona fide viral genome associated proteins, we searched the Contaminant Repository for Affinity Purification (CRAPome) [36] for cellular proteins identified by these methods (Fig F in S1 Text). This web-based database includes 411 datasets of common contaminants present in negative controls for protein purification. Most proteins that were identified in this study were found in less than 20% of the negative control datasets, consistent with specific enrichment of viral genome associated proteins by these methods.
Taken together, aniPOND is an alternative method for the purification of virus-associated proteins and may be more useful in situations were few genomes are present (for example before DNA replication) or when genomes are not efficiently labeled with nucleoside analogs (for example wild type KOS). Furthermore, the combination of both methods reveals a comprehensive look at proteins associated with viral genomes.
Cellular factors relocalize to viral replication compartments during HSV infection
To better visualize the reorganization of host nuclear factors to viral replication compartments during lytic infection with HSV, we used immunofluorescence to compare the distribution of cellular factors in the nucleus of mock-infected cells to cells infected with KOS for 8 hours (Fig 5). Ten cellular proteins that were identified by iPOND and/or aniPOND, including replication proteins PCNA and TOP2, transcription factors TFII-I, Spt5, Spt6, and XPD, chromatin remodeling factors SSRP1, HMGB1, and HDAC2, as well as the repair protein Ku70 were tested for colocalization with viral DNA. In all cases, the cellular proteins relocalized to viral replication compartments. Interestingly, these cellular factors were relocated from multiple locations within the nucleus. Taken together, it is clear that HSV infection induces gross reorganization of the host nucleus and compartmentalization of cellular factors that likely participate in multiple aspects of the virus life cycle.

HSV genomes are deficient for histones during lytic infection
Micrococcal nuclease digestion assays indicate that packaged genomes are not associated with nucleosomes, only a small portion of incoming unreplicated genomes are associated with nucleosomes, and newly replicated genomes are not associated with nucleosomes [37–39]. However, ChIP mapping data indicate that histones are bound to many HSV promoters and genes, and often have marks of active chromatin [40,41]. The working model is that histones are present on viral genomes during early lytic infection, the distribution and density of histones on lytic genomes is significantly less than the host genome, and histones likely play a role in the regulation of viral gene expression. In contrast, latent genomes are associated with ordered chromatin similar to host cell DNA [42].
Many components of chromatin remodeling complexes were identified on replicated viral genomes by iPOND and aniPOND (Table 4). These include members of the FACT, INO80, NURD, and SWI/SNF chromatin remodeling complexes, as well as DNA and chromatin modifying enzymes. However, histones were not enriched on purified replicated genomes, with the exceptions of a few histone H1 variants, which were also abundant in negative controls (Table 4 and Fig F in S1 Text). Perhaps chromatin remodeling factors associate with viral DNA to facilitate the removal of histones or to keep histones from binding to newly replicated genomes.
To provide support for the absence of histones on replicating genomes, colocalization of viral genomes with histones was assayed by fluorescence microscopy. EdU-labeled viral replication compartments were tagged with Alexa Fluor 488 and either histone H1 (all subtypes; Fig 6A) or H3 (6B) was labeled with specific antibodies for immunofluorescence. Less dense localization of both histones was observed with viral DNA relative to cellular DNA. This localization pattern greatly contrasts the pattern observed for proteins that were identified to associate with viral genomes (Fig 5). These data support iPOND and aniPOND results and confirm that histones are not enriched on viral genomes during DNA replication.

To assay for the colocalizaiton of viral genomes with histones during early lytic infection, fluorescence imaging of prelabeled incoming viral genomes was carried out at 2 hpi (Fig 7). Histones H1 and H3 did not colocalize with incoming viral genomes, at least within limits of detection by immunofluorescence. This is in stark contrast to the pattern of ICP4 colocalization with incoming genomes. In conclusion, iPOND, aniPOND, and imaging data provide support for a deficiency of histones on viral genomes throughout lytic infection

Discussion
In this study, we adapted procedures that have been used to label and purify cellular replication forks to label and purify replicating HSV-1 genomes. We have engineered mutant HSV strains that increase EdU incorporation into replicating viral genomes allowing for more sensitive imaging and purification of viral DNA. We are the first to label HSV DNA to track the fate of incoming viral genomes within an infected cell, demonstrating its colocalization with ICP4 expressed from those genomes. We have optimized the iPOND and aniPOND methods to study HSV genomes during different stages of the HSV life cycle by these methods. From these studies we have compiled a comprehensive list of proteins that are selectively recruited to HSV genomes during and after viral DNA replication. By imaging the relocalization of several of these factors to viral replication compartments during infection, we have demonstrated the extent to which host nuclei are largely reorganized during viral infection. Finally, we show that viral genomes isolated by iPOND and aniPOND have a relatively low abundance of histones, which is consistent with the lack of colocalization of genomes with histones H1 and H3. Data presented in this paper provide a comprehensive view of viral and cellular proteins that associate with replicating HSV genomes during productive infection and provide insight into how HSV manipulates host cell nuclear machineries for the expression, replication, and maintenance of its genome.
In this study, we identified >200 viral and cellular proteins that are associated with HSV genomes after the onset of DNA replication. The overall most abundant proteins found on the genome include the viral transcription factor ICP4 (Table 3), and the viral replication proteins UL29 (ICP8, major DNA binding protein), UL30 (viral DNA polymerase), and UL42 (processivity factor) (Table 2). The identified cellular proteins function in host cell nuclear processes including DNA replication, repair, chromatin remodeling, transcription, RNA processing, and nuclear transport (Figs 3C and 4C).
Viral DNA replication
In addition to UL29, UL30, and UL42, four other viral replication factors, UL9 (origin binding protein) and UL5/UL8/UL52 (helicase primase complex) were enriched on viral genomes. In contrast to iPOND studies of cellular replication forks [23,26,28], most cellular DNA replication proteins did not copurify with viral genomes. However, the cellular processivity factor PCNA and topoisomerases TOP1, TOP2a, and TOP2b were reproducibly enriched on replicating viral genomes. iPOND data indicate that the levels of PCNA on viral genomes is higher at 6 hpi than at 8 and 12 hpi, suggesting that PCNA may play a role in early phases of viral DNA replication. Topoisomerases are important for relaxing supercoiled DNA as a consequence of helicase unwinding during replication and transcription [43], and likely carryout this same function on viral DNA. PCNA (Fig 5)[44] and Top2 (Fig 5) redistribute to viral replication compartments during viral DNA replication, however a direct role in HSV replication has yet to be demonstrated. Currently, there is not a good system to study origin-primed viral DNA replication in vitro [20]. It is possible that cellular PCNA or topoisomerases are the missing players in these reconstitution assays.
The DNA damage response, repair, and recombination
We also identified several components involved in double strand break (DSB) recognition and repair associated with replicating viral genomes in our assays. These include Ku70 and Ku80, the Mre11/Rad50/Nbs1 (MRN) complex, ataxia telangiectasia mutated (ATM), and the catalytic subunit of DNA dependent protein kinase (DNA-PKcs) (Table 5). Ku70 (Fig 5) and Ku80 [45] colocalize with viral replication compartments. However, Ku70 expression is inhibitory for viral DNA replication [45]. Perhaps, these proteins participate in a cellular antiviral response in attempt to control virus multiplication.
The MRN complex, ATM, and activation of the DNA damage response are beneficial for HSV genome replication [46–48]. The MRN complex and ATM are recruited to viral replication compartments and ATM is activated through autophosphorylation to trigger the DNA damage response and cell cycle arrest through multiple pathways. In this way, the cell recognizes the viral genome as DNA damage. However, downstream binding of cellular proteins that mediate repair through nonhomologous end joining (NHEJ) and homologous recombination (HR) pathways are inhibited by the actions of the viral E3 ubiquitin ligase, ICP0 [49–51]. ICP0 targets downstream factors in these double strand break repair pathways for degradation, including DNA-PKcs, RNF8, and RNF168. Consistent with these data, we did not identify RNF8 or RNF168 to be recruited to viral DNA in our assays. Purification of DNA-PKcs is not inconsistent with these observations because only 50% of DNA-PKcs is degraded by ICP0 and this is likely cell type specific [45]. These data support a scenario whereby viral genomes trigger the DNA damage response and cell cycle arrest to create an environment that is conducive to viral DNA replication. ICP0 may inhibit the actions of cellular HR and NHEJ pathways for the repair of virus ends, as well as nicks and gaps that occur during viral DNA replication. It is possible that HSV-1 instead uses its own machinery for recombination and repair during DNA replication, mediated by the actions of ICP8 and UL12 (alkaline nuclease) [52,53]. In fact, UL12 has been shown to interact with components of the MRN complex and may therefore act with the MRN complex to carryout virus specific recombination [54].
The structural maintenance of chromosomes (SMC) family of ATPases function to stabilize and organize chromosomes during mitosis [55]. Of these complex members, SMC1 and SMC3, which make up the core of the cohesion complex, reproducibly copurify with replicating viral genomes. The cohesion complex is essential for sister chromatid cohesion during mitosis, but also plays a role in transcription and DNA repair by recombination [56]. Cohesin complex proteins SMC3 and Rad21 have previously been shown to associate with Epstein-Barr virus genomes [57,58]. Perhaps these proteins are involved in HSV gene expression or recombination during DNA replication. Mismatch repair [59] and base excision repair [60] pathways also function in maintaining HSV genomes, and specific factors involved in both of these types of repair were found to be associated with viral genomes in this study (Table 5).
RNA processing
RNA processing factors involved in all steps in pre-mRNA processing including capping, splicing, polyadenylation, and export were abundant on viral genomes (Table 6). Interestingly, ICP27, an essential viral immediate early gene product that regulates all steps in viral RNA processing [61] was not readily detectable on genomes. However, the TREX complex was found in our studies, which has been shown to interact with ICP27 [62,63] and to be involved in the export of KSHV intronless mRNAs [64]. RNA helicases, which are involved in all aspects of RNA metabolism, as well as components of the nuclear transport machinery were also found associated with viral genomes. The abundant isolation of all of these RNA processing factors is most likely consistent with the high level accumulation of viral mRNA late after infection and the fact that transcription and RNA processing are coupled [65–67].
Chromatin remodeling and transcription
Multiple components of several chromatin remodeling complexes were enriched on viral genomes including the FACT, INO80, NURD, and SWI/SNF complexes (Table 4). This is consistent with proteomic analysis of proteins bound to ICP4 extracted from virus infected cells, in which components of INO80, NURD, and SWI/SNF complexes were identified [34]. One of the FACT complex members, SPT16, was shown to copurify with ICP8 in the absence of DNAse treatment [45] and here we demonstrated the redistribution of the other FACT complex member SSRP1 to viral replication compartments (Fig 5). As discussed above, histones were not enriched on viral genomes, raising the possibility that these complexes maintain a nucleosome or histone free state, greatly facilitating processes such as replication and transcription on the genome. The FACT complex has been shown to disrupt nucleosome structure and allow DNA and RNA polymerases to access the DNA [68], the INO80 complex mediates nucleosome sliding [69], the NURD complex has both histone deacetylase and nucleosome remodeling functions [70], and high mobility group (HMG) proteins, which are also found on viral genomes, have been shown to increase accessibility of chromatin-bound DNA [71]. Furthermore, the INO80 and FACT complexes have also been implicated in cellular DNA damage repair by homologous recombination [72,73], and may therefore also play roles in mechanisms of viral DNA recombination. HMGB1 was previously shown to function as a coactivator for ICP4 mediated transcription in vitro [74], and may therefore function to mediate promoter specific activation of viral genes.
RNA polymerase II (polII) was abundant on isolated viral genomes (Table 3), with RPB1 and RPB2 being the most enriched subunits, most likely because they make direct contact with DNA during transcription [75]. The transcription elongation factors Spt5, Spt6 [76], and Trim28 [77] were also found associated with viral genomes and Spt5 and Spt6 were shown to relocalize to viral replication compartments (Fig 5). These are therefore likely candidates to regulate elongation during HSV transcription. TFII-I binds to initiator (inr) elements in cellular promoters [78] and therefore may play a role in the expression of late viral genes.
The viral transcriptional regulators VP16, ICP4, and ICP22 were found on viral genomes by both iPOND and aniPOND. ICP22 was previously found to associate with ICP4 and RNA polII in transcription complexes [79] and to mediate phosphorylation of polII [80]. VP16 is a tegument protein that activates transcription of immediate early viral genes [11]. ICP4 regulates expression from early and late HSV promoters and repression of immediate early promoters. It interacts with TFIID, TFIIH, and a specific form of the mediator complex that lacks Med26 and contains the kinase domain [34,35]. Here we show that viral genomes copurified with subunits of TFIID, TFIIH, and the same form of the mediator complex that copurified with ICP4 from Vero [34] and resting MRC-5 cells (Table B in S1 Text). This form of mediator possesses the kinase domain, but lacks med26, and thus may be involved in repression, possibly of immediate early promoters late after infection. The viral genes transcribed late after infection all possess relatively simple TATA box-containing promoters, yet are abundantly transcribed. The accumulated data support a model where ICP4 plays an integral role in recruiting most of the key polII transcription factors, such as TFIID, required for abundant late transcription.
This study has provided a comprehensive view of the viral and cellular proteins associated with replicating HSV genomes and provides new insight into cellular mechanisms that regulate HSV infection. The presence of cellular proteins involved in a variety of nuclear processes is consistent with the rapid and high level of accumulation of viral transcripts, replicated genomes, and progeny virions shortly after infection. This must be accompanied by the recombination and repair of replicating genomes. It is probable that the association and function of these factors is facilitated by the relative dearth of cellular chromatin, which may be a function of the recruitment of multiple chromatin remodeling complexes. In this model, ICP4 binding to the genome, may have multiple roles in recruiting chromatin remodeling complexes and key polII transcription complexes, although a direct role of ICP4 in chromatin organization has yet to be demonstrated. What remains to be studied is to what extent this state is determined prior to the onset of viral DNA replication.
Materials and Methods
Cells and viruses
Experiments were performed using MRC-5 (human embryonic lung) or Vero (African green monkey kidney) cells obtained from and propagated as recommended by ATCC. The viruses used in this study include the wild type HSV-1 strain, KOS, as well as UL2, UL50, and UL2/UL50 mutant viruses. Mutants were generated in bacterial artificial chromosomes (BACs) containing full-length, infectious KOS DNA [81] using two-step red-mediated recombination [82,83]. To generate the UL2 null virus, the cassette GGCTAGTTAACTAGCC, which contains a premature termination codon in all three reading frames, as well as an HpaI restriction site for validation, was inserted after the codon for cysteine 75 of the UL2 open reading frame. For the UL50 null virus, the codon for alanine 110 was replaced with this cassette. Mutant KOS-BAC constructs were transfected using Lipofectamine 2000 Transfection Reagent (Life Technologies) and propagated in Vero cells. Viral DNA was isolated from individual plaques [84] and screened for mutations by Southern blotting [85].
Preparation of prelabeled viral genomes
To generate prelabeled virus stocks, 1x108 Vero cells were infected with unlabeled KOS or UL2/UL50 at an MOI of 10 PFU/cell at 37°C for 1 hour. After rinsing with tris-buffered saline (TBS) to remove unadsorbed virus, media was replaced with Dulbecco’s Modified Eagle Medium (DMEM) containing 5% fetal bovine serum (FBS). Four hpi, EdU (Sigma-Aldrich) was added to the growth medium at the indicated concentration and incubated for an additional 34–36 hours. Monolayers were harvested, freeze-thawed three times at -80°C, sonicated, and clarified by low-speed centrifugation. Viral titers were determined by plaque assay on Vero cells.
Click chemistry and immunofluorescence
A total of 2x105 Vero cells were grown on glass coverslips in 12-well dishes. Infections were carried out at an MOI of 10 in 100 μl TBS for 1 hour at room temperature. After infection, inoculum was removed and cells were rinsed with 1 ml TBS prior to addition of 1 ml DMEM plus 5% FBS. Infections were carried out at 37°C for the indicated period of time, with or without the addition of EdU to the growth medium. Cells were fixed with 3.7% formaldehyde for 15 min, washed two times with phosphate-buffered saline (PBS), permeabilized with 0.5% Triton-X 100 for 20 min, and blocked with 3% bovine serum albumin (BSA) for 30 min. EdU-labeled DNA was conjugated to Alexa Fluor 488 azide using the Click-iT EdU imaging kit according to manufacturer’s protocol (Life Technologies). Cells were rinsed with PBS plus 3% BSA, then PBS, labeled with Hoechst 33342 (1 : 2000 dilution) for 30 min, washed two times with PBS, then incubated with primary antibody (mouse anti-ICP4 : 58S, 1 : 500; mouse anti-histone H1: ab4269 (Abcam), 1 : 1000; rabbit anti-histone H3: ab1791 (Abcam), 1 : 1000; mouse anti-PCNA: sc-056 (Santa Cruz), 1 : 200; mouse anti-topoisomerase 2: NA14 (Calbiochem), 1 : 20; goat anti-TFII-I: sc-9943x (Santa Cruz), 1 : 200; rabbit anti-Spt5: A300-869A (Bethyl Laboratories), 1 : 200; rabbit anti-Spt6: ab32820 (Abcam), 1 : 200; rabbit anti-TFIIH p89 (XPD): sc-293 (Santa Cruz), 1 : 200; mouse anti-SSRP1 : 10D1 (Biolegend), 1 : 200; rabbit anti-HMGB1: ab18256 (Abcam), 1 : 1000; rabbit anti-HDAC2: sc-7899 (Santa Cruz), 1 : 200; rabbit anti-Ku70: sc-9033 (Santa Cruz), 1 : 200) and Alexa Fluor 594-conjugated secondary antibodies (Santa Cruz, 1 : 500) as described previously [34]. Images were obtained using an Olympus Fluoview FV1000 confocal microscope.
iPOND
iPOND was carried out as described previously [25] with the following modifications. For each condition, three 500 cm2 tissue culture dishes containing confluent monolayers of MRC-5 cells (~7x107 cells/dish) were infected with UL2/UL50 double mutant virus at an MOI of 10 PFU/cell for one hour at room temperature. After adsorption, the inoculum was removed and cells were rinsed with TBS before addition of fresh DMEM plus 5% FBS. Cells were incubated at 37°C for the indicated period of time before addition of EdU at a final concentration of 2.5 μM. After incubation for an additional 2–4 hours, cells were fixed with 1% (wt/vol) formaldehyde in PBS for 15 min at room temperature, quenched with 125 mM glycine, and harvested by scraping. Cell permeabilization, click chemistry, cell lysis, sonication, and streptavidin capture were carried out as described except the samples were sonicated 6 times for 30 sec each at 7 watts using a Cole-Palmer ultrasonic processor with microtip. For each condition, samples from three plates were combined, and proteins were eluted from streptavidin-coated beads by boiling in 200 μl 2x SDS Laemmli sample buffer to reverse formaldehyde crosslinks.
aniPOND
aniPOND was carried out as described previously [33] with the following modifications. For each condition, one 500 cm2 tissue culture dish containing a confluent monolayer of MRC-5 cells (~7x107 cells) was infected with wild type KOS or UL2/UL50 double mutant virus at an MOI of 10 PFU/cell for one hour at room temperature. After adsorption, the inoculum was removed and cells were rinsed with TBS before addition of fresh DMEM plus 5% FBS. Cells were incubated at 37°C for four hours before the addition of EdU (Sigma-Aldrich) at a final concentration of 2.5 μM, followed by an additional four-hour incubation. To detach the monolayer and extract nuclei, 20 ml nuclear extraction buffer was added directly to each plate, incubated at 4°C for 15 min, and harvested by scraping. Cell washes, click chemistry, cell lysis, sonication, and streptavidin capture were carried out as described except for cell lysis cells were incubated for 30 min total in lysis buffer and sonicated 8 times for 30 sec each at 7 watts. Proteins were eluted from streptavidin-coated beads by boiling in 66 μl 2x SDS Laemmli sample buffer. For aniPOND experiment 2, two plates of MRC-5 cells were used for analysis and for protein elution, streptavidin-coated beads from both samples were combined and proteins were eluted in 66 μl 2x sample buffer to generate a 2x concentrated sample.
Western blotting
SDS polyacrylamide gel electrophoresis and western blotting were carried out as described previously [86]. Proteins were transferred to polyvinylidine fluoride membranes (Amersham) for chemi-luminescent detection with ECL reagent (Amersham). For detection of ICP4, membranes were probed with the 58S polyclonal mouse antibody (1 : 5000 dilution).
Mass spectrometry and data analysis
Mass spectrometry was carried out by MSBioworks. The entire sample was separated ~1.5cm on a 10% Bis-Tris Novex mini-gel (Invitrogen) using the MES buffer system. The gel was stained with coomassie and excised into ten equally sized segments. Gel segments were processed as described and analyzed by nano liquid chromatography with tandem mass spectrometry (LC/MS/MS) [87]. Data were searched using Mascot and Mascot DAT files were parsed into the Scaffold software for validation, filtering, and to create a nonredundant list per sample. Data were filtered at 1% protein and peptide level false discovery rates and requiring at least two unique peptides per protein. Proteins were considered most significantly enriched by iPOND or aniPOND based on the following criteria: 1) protein had at least 5 spectral counts (SpC) in the experimental sample, 2) protein was not detected in the control or was enriched over the control by at least four-fold based on dividing SpC values, and 3) was detected in duplicate experiments. Raw SpC data without normalization are presented in Tables 1–6 and Table A in S1 Text.
DNA isolation and quantitative real-time PCR (qRT-PCR)
DNA was isolated from 1/20th volume cell lysates or 1/10th volume streptavidin-coated beads during iPOND and aniPOND experiments. For isolation of DNA from cell lysates, an equal volume of 2x SDS-bicarb solution (2% SDS, 0.2 M NaHCO3) was added to the sample and for isolation of bead-bound DNA, beads were resuspended in 1x SDS-bicarb solution. Samples were incubated at 65°C overnight, followed by extraction with phenol:chloroform:isoamyl alcohol (25 : 24 : 1) and chloroform:isoamyl alcohol (24 : 1). DNA was recovered using the MinElute PCR Purification kit (Qiagen). DNA concentrations were measure using a Quibit Fluorometer and the Qubit dsDNA HS Assay Kit (Life Technologies).
qRT-PCR was carried out as described previously [88]. The HSV-1 TK gene was amplified to estimate the amount of viral DNA in each sample. Primers used for amplification of the HSV TK gene were TkdsF1 (5´-ACCCGCTTAACAGCGTCAACA-3´) and TkdsR1 (5´-CCAAAGAGGTGCGGGAGTTT-3´). Standard curves were generated using purified KOS DNA.
Supporting Information
Zdroje
1. McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, et al. (1988) The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69 : 1531–1574. 2839594
2. McGeoch DJ, Dolan A, Donald S, Brauer DH (1986) Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res 14 : 1727–1745. 3005980
3. Batterson W, Furlong D, Roizman B (1983) Molecular genetics of herpes simplex virus. VIII. further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol 45 : 397–407. 6296445
4. Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A (2000) Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20 : 4922–4931. 10848617
5. Shahin V, Hafezi W, Oberleithner H, Ludwig Y, Windoffer B, et al. (2006) The genome of HSV-1 translocates through the nuclear pore as a condensed rod-like structure. J Cell Sci 119 : 23–30. 16339172
6. Maul GG (1998) Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20 : 660–667. 9780840
7. Smith S, Reuven N, Mohni KN, Schumacher AJ, Weller SK (2014) Structure of the herpes simplex virus 1 genome: manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J Virol 88 : 10146–10156. doi: 10.1128/JVI.01723-14 24965466
8. Everett RD (2001) DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20 : 7266–7273. 11704855
9. Taylor TJ, McNamee EE, Day C, Knipe DM (2003) Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology 309 : 232–247. 12758171
10. Batterson W, Roizman B (1983) Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J Virol 46 : 371–377. 6302308
11. Campbell ME, Palfreyman JW, Preston CM (1984) Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J Mol Biol 180 : 1–19. 6096556
12. Watson RJ, Clements JB (1980) A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285 : 329–330. 6246451
13. Alwine JC, Steinhart WL, Hill CW (1974) Transcription of herpes simplex type 1 DNA in nuclei isolated from infected HEp-2 and KB cells. Virology 60 : 302–307. 4366499
14. Everett RD, Sourvinos G, Orr A (2003) Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J Virol 77 : 3680–3689. 12610143
15. Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A (2004) Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J Virol 78 : 1903–1917. 14747555
16. La Boissiere S, Izeta A, Malcomber S, O'Hare P (2004) Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J Virol 78 : 8002–8014. 15254172
17. Honess RW, Roizman B (1974) Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14 : 8–19. 4365321
18. Honess RW, Roizman B (1975) Regulation of herpesvirus macromolecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc Natl Acad Sci U S A 72 : 1276–1280. 165503
19. Challberg MD (1986) A method for identifying the viral genes required for herpesvirus DNA replication. Proc Natl Acad Sci U S A 83 : 9094–9098. 3024166
20. Weller SK, Coen DM (2012) Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol 4: a013011. doi: 10.1101/cshperspect.a013011 22952399
21. Cardone G, Heymann JB, Cheng N, Trus BL, Steven AC (2012) Procapsid assembly, maturation, nuclear exit: dynamic steps in the production of infectious herpesvirions. Adv Exp Med Biol 726 : 423–439. doi: 10.1007/978-1-4614-0980-9_19 22297525
22. Wang IH, Suomalainen M, Andriasyan V, Kilcher S, Mercer J, et al. (2013) Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 14 : 468–480. doi: 10.1016/j.chom.2013.09.004 24139403
23. Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, et al. (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288 : 31458–31467. doi: 10.1074/jbc.M113.511337 24047897
24. Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, et al. (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25 : 1320–1327. doi: 10.1101/gad.2053211 21685366
25. Sirbu BM, Couch FB, Cortez D (2012) Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7 : 594–605. doi: 10.1038/nprot.2012.010 22383038
26. Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, et al. (2013) A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep 3 : 1105–1116. doi: 10.1016/j.celrep.2013.03.009 23545495
27. Dungrawala H, Cortez D (2015) Purification of proteins on newly synthesized DNA using iPOND. Methods Mol Biol 1228 : 123–131. doi: 10.1007/978-1-4939-1680-1_10 25311126
28. Aranda S, Rutishauser D, Ernfors P (2014) Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Res 42 : 6972–6986. doi: 10.1093/nar/gku374 24852249
29. Everett RD, Murray J, Orr A, Preston CM (2007) Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81 : 10991–11004. 17670833
30. Hobbs WE, DeLuca NA (1999) Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. Journal of Virology 73 : 8245–8255. 10482575
31. Lomonte P, Everett RD (1999) Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J Virol 73 : 9456–9467. 10516054
32. Haug T, Skorpen F, Aas PA, Malm V, Skjelbred C, et al. (1998) Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res 26 : 1449–1457. 9490791
33. Leung KH, Abou El Hassan M, Bremner R (2013) A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 55 : 204–206. doi: 10.2144/000114089 24107252
34. Wagner LM, DeLuca NA (2013) Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in PolII transcription. PLoS One 8: e78242. doi: 10.1371/journal.pone.0078242 24147125
35. Lester JT, DeLuca NA (2011) Herpes simplex virus 1 ICP4 forms complexes with TFIID and mediator in virus-infected cells. J Virol 85 : 5733–5744. doi: 10.1128/JVI.00385-11 21450820
36. Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, et al. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10 : 730–736. doi: 10.1038/nmeth.2557 23921808
37. Leinbach SS, Summers WC (1980) The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J Gen Virol 51 : 45–59. 6257837
38. Mouttet ME, Guetard D, Bechet JM (1979) Random cleavage of intranuclear herpes simplex virus DNA by micrococcal nuclease. FEBS Lett 100 : 107–109. 220083
39. Oh J, Fraser NW (2008) Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol 82 : 3530–3537. 18160436
40. Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, et al. (2004) During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol 78 : 10178–10186. 15331750
41. Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, et al. (2006) Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J Virol 80 : 5740–5746. 16731913
42. Deshmane SL, Fraser NW (1989) During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63 : 943–947. 2536115
43. Capranico G, Marinello J, Baranello L (2010) Dissecting the transcriptional functions of human DNA topoisomerase I by selective inhibitors: implications for physiological and therapeutic modulation of enzyme activity. Biochim Biophys Acta 1806 : 240–250. doi: 10.1016/j.bbcan.2010.06.003 20600630
44. Wilcock D, Lane DP (1991) Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349 : 429–431. 1671528
45. Taylor TJ, Knipe DM (2004) Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol 78 : 5856–5866. 15140983
46. Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD (2005) DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102 : 5844–5849. 15824307
47. Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, et al. (2005) Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem 280 : 30336–30341. 15964848
48. Lilley CE, Schwartz RA, Weitzman MD (2007) Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15 : 119–126. 17275307
49. Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, et al. (2010) A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J 29 : 943–955. doi: 10.1038/emboj.2009.400 20075863
50. Parkinson J, Lees-Miller SP, Everett RD (1999) Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73 : 650–657. 9847370
51. Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD (2011) The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7: e1002084. doi: 10.1371/journal.ppat.1002084 21698222
52. Wilkinson DE, Weller SK (2003) The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 55 : 451–458. 14609200
53. Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, et al. (2012) The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog 8: e1002862. doi: 10.1371/journal.ppat.1002862 22912580
54. Balasubramanian N, Bai P, Buchek G, Korza G, Weller SK (2010) Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J Virol 84 : 12504–12514. doi: 10.1128/JVI.01506-10 20943970
55. Hirano T (2005) SMC proteins and chromosome mechanics: from bacteria to humans. Philos Trans R Soc Lond B Biol Sci 360 : 507–514. 15897176
56. Mehta GD, Kumar R, Srivastava S, Ghosh SK (2013) Cohesin: functions beyond sister chromatid cohesion. FEBS Lett 587 : 2299–2312. doi: 10.1016/j.febslet.2013.06.035 23831059
57. Chen HS, Martin KA, Lu F, Lupey LN, Mueller JM, et al. (2014) Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol 88 : 1703–1713. doi: 10.1128/JVI.02209-13 24257606
58. Holdorf MM, Cooper SB, Yamamoto KR, Miranda JJ (2011) Occupancy of chromatin organizers in the Epstein-Barr virus genome. Virology 415 : 1–5. doi: 10.1016/j.virol.2011.04.004 21550623
59. Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD (2011) DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J Virol 85 : 12241–12253. doi: 10.1128/JVI.05487-11 21957315
60. Mullaney J, Moss HW, McGeoch DJ (1989) Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase. J Gen Virol 70 (Pt 2): 449–454. 2567340
61. Sandri-Goldin RM (2011) The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6 : 1261–1277. doi: 10.2217/fmb.11.119 22082288
62. Chen IH, Li L, Silva L, Sandri-Goldin RM (2005) ICP27 recruits Aly/REF but not TAP/NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J Virol 79 : 3949–3961. 15767397
63. Chen IH, Sciabica KS, Sandri-Goldin RM (2002) ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J Virol 76 : 12877–12889. 12438613
64. Boyne JR, Colgan KJ, Whitehouse A (2008) Recruitment of the complete hTREX complex is required for Kaposi's sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog 4: e1000194. doi: 10.1371/journal.ppat.1000194 18974867
65. Montes M, Becerra S, Sanchez-Alvarez M, Sune C (2012) Functional coupling of transcription and splicing. Gene 501 : 104–117. doi: 10.1016/j.gene.2012.04.006 22537677
66. Pandit S, Wang D, Fu XD (2008) Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol 20 : 260–265. doi: 10.1016/j.ceb.2008.03.001 18436438
67. Luna R, Gaillard H, Gonzalez-Aguilera C, Aguilera A (2008) Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma 117 : 319–331. doi: 10.1007/s00412-008-0158-4 18427828
68. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, et al. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301 : 1090–1093. 12934006
69. Jin J, Cai Y, Yao T, Gottschalk AJ, Florens L, et al. (2005) A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. J Biol Chem 280 : 41207–41212. 16230350
70. Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL (1998) Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395 : 917–921. 9804427
71. Ozturk N, Singh I, Mehta A, Braun T, Barreto G (2014) HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol 2 : 5. doi: 10.3389/fcell.2014.00005 25364713
72. Wu S, Shi Y, Mulligan P, Gay F, Landry J, et al. (2007) A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol 14 : 1165–1172. 18026119
73. Oliveira DV, Kato A, Nakamura K, Ikura T, Okada M, et al. (2014) Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J Cell Sci 127 : 763–772. doi: 10.1242/jcs.135855 24357716
74. Carrozza MJ, DeLuca N (1998) The high mobility group protein 1 is a coactivator of herpes simplex virus ICP4 in vitro. J Virol 72 : 6752–6757. 9658123
75. Kettenberger H, Armache KJ, Cramer P (2003) Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell 114 : 347–357. 12914699
76. Kwak H, Lis JT (2013) Control of transcriptional elongation. Annu Rev Genet 47 : 483–508. doi: 10.1146/annurev-genet-110711-155440 24050178
77. Bunch H, Zheng X, Burkholder A, Dillon ST, Motola S, et al. (2014) TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat Struct Mol Biol 21 : 876–883. doi: 10.1038/nsmb.2878 25173174
78. Roy AL, Meisterernst M, Pognonec P, Roeder RG (1991) Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature 354 : 245–248. 1961251
79. Leopardi R, Ward PL, Ogle WO, Roizman B (1997) Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 proteinkinase. J Virol 71 : 1133–1139. 8995634
80. Lin FS, Ding Q, Guo H, Zheng AC (2010) The herpes simplex virus type 1 infected cell protein 22. Virol Sin 25 : 1–7. doi: 10.1007/s12250-010-3080-x 20960278
81. Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, et al. (2006) Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135 : 197–206. 16647145
82. Tischer BK, von Einem J, Kaufer B, Osterrieder N (2006) Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40 : 191–197. 16526409
83. Tischer BK, Smith GA, Osterrieder N (2010) En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634 : 421–430. doi: 10.1007/978-1-60761-652-8_30 20677001
84. Samaniego LA, Webb AL, DeLuca NA (1995) Functional interactions between herpes simplex virus immediate-early proteins during infection: gene expression as a consequence of ICP27 and different domains of ICP4. J Virol 69 : 5705–5715. 7637016
85. Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98 : 503–517. 1195397
86. Wagner LM, Lester JT, Sivrich FL, DeLuca NA (2012) The N terminus and C terminus of herpes simplex virus 1 ICP4 cooperate to activate viral gene expression. J Virol 86 : 6862–6874. doi: 10.1128/JVI.00651-12 22496239
87. Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, et al. (2012) Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci 32 : 3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012 22423091
88. Harkness JM, Kader M, DeLuca NA (2014) Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J Virol 88 : 6847–6861. doi: 10.1128/JVI.00516-14 24719411
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance