
A Central Role for Carbon-Overflow Pathways in the Modulation of Bacterial Cell Death
Many bacterial species including the pathogen Staphylococcus aureus are capable of adhering to surfaces and forming complex communities called biofilms. This mode of growth can be particularly challenging from an infection control standpoint, as they are often refractory to antibiotics and host immune system. Although developmental processes underlying biofilm formation are not entirely clear, recent evidence suggests that cell death of a subpopulation is crucial for its maturation. In this study we provide insight regarding the metabolic pathways that control cell death and demonstrate that acetate, a by-product of glucose catabolism, potentiates a form of cell death that exhibits physiological and biochemical hallmarks of apoptosis in eukaryotic organisms. Finally, we demonstrate that altering the ability of metabolic pathways that regulate acetate mediated cell death in S. aureus affects the outcome of biofilm-related diseases, such as infective endocarditis.
Published in the journal:
A Central Role for Carbon-Overflow Pathways in the Modulation of Bacterial Cell Death. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004205
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004205
Summary
Many bacterial species including the pathogen Staphylococcus aureus are capable of adhering to surfaces and forming complex communities called biofilms. This mode of growth can be particularly challenging from an infection control standpoint, as they are often refractory to antibiotics and host immune system. Although developmental processes underlying biofilm formation are not entirely clear, recent evidence suggests that cell death of a subpopulation is crucial for its maturation. In this study we provide insight regarding the metabolic pathways that control cell death and demonstrate that acetate, a by-product of glucose catabolism, potentiates a form of cell death that exhibits physiological and biochemical hallmarks of apoptosis in eukaryotic organisms. Finally, we demonstrate that altering the ability of metabolic pathways that regulate acetate mediated cell death in S. aureus affects the outcome of biofilm-related diseases, such as infective endocarditis.
Introduction
The balanced progression of cell division and apoptotic events is a classic hallmark of eukaryotic development [1]. Intriguingly, a similar homeostatic control of cell death, lysis and proliferation is predicted to benefit the development of adherent multicellular bacterial assemblages (called biofilms) by providing nutrients and critical biofilm building matrix components like extracellular DNA (eDNA) [2], [3]. Consistent with this assumption, recent investigations have revealed that bacteria, like eukaryotes not only harbor elaborate regulatory systems that modulate cell death, but also display biochemical and physiological hallmarks characteristic of programmed cell death (PCD) [4], [5], [6].
The molecular components that mediate cell death in S. aureus are regulated, in part, by the LysR-type transcriptional regulator, CidR [7] and include a set of membrane bound proteins, CidA and CidB, whose functions are predicted to be analogous to the Bcl-2 family of apoptotic modulators in eukaryotes [2], [3]. However, less clear are the mechanistic contributions of other members of the CidR regulon in cell death, specifically those enzymes that are active during overflow metabolism, pyruvate oxidase (CidC) and α-acetolactate synthase/decarboxylase (AlsSD) [8], [9]. Given that these enzymes are the only additional members of the CidR regulon and that multiple physiological signals that directly affect both central metabolism and cell senescence coordinate their expression [10], we predicted an intricate role for these proteins in the physiology of cell death.
Here, we report that both CidC and AlsSD carbon-overflow pathways contribute to staphylococcal cell death. Our results demonstrate that cell death is potentiated by acetate, a major weak acid byproduct of glucose catabolism, whose levels are antithetically modulated by CidC and AlsSD activities. We also report that the physiological features accompanying staphylococcal cell death resemble eukaryotic PCD (apoptosis) wherein cell death is associated with respiratory dysfunction, increased ROS production and DNA damage. Finally, we demonstrate a role for staphylococcal PCD in biofilm development and pathogenesis.
Results
Glucose-dependent cell death in stationary phase exhibits hallmarks of PCD
Multiple studies have linked the uptake and metabolic fate of glucose to the regulation of PCD during eukaryotic development [11]. To determine whether such correlations are broadly conserved in bacteria, the effects of glucose on staphylococcal cell death were assessed over a period of five days by monitoring the colony forming units (cfu/ml) of wild-type cells grown aerobically in rich media (tryptic soy broth, TSB) containing either 14 mM or 35 mM glucose. Although there appeared to be no significant difference in the viable cell counts after 24 h of growth in either type of media, subsequent stationary phase survival of wild-type cells was dependent on initial glucose concentrations wherein S. aureus grown in TSB-35 mM glucose displayed a steep decline in viability (∼7 log10 difference) compared to the modest decline (∼1.2 log10) observed for cells grown in TSB-14 mM glucose over the same period of time (120 h) (Fig. 1A). These results indicate that growth in excess glucose reduced the survival of S. aureus in stationary phase.

To ascertain whether excess glucose-mediated staphylococcal cell death bore similarities to PCD, we explored the physiological status of the dying population by flow cytometry after 72 h of growth and compared them to a 24 h reference point when cells were relatively healthy based on viable counts. Respiratory potential was estimated using the cell permeable redox dye, cyano-2,3-ditolyl tetrazolium chloride (CTC). Reduction of CTC into a red insoluble fluorescent formazan that accumulates intracellularly is achieved by dehydrogenases of the electron transport chain (ETC) that are expressed within actively respiring bacterial populations [12]. In addition to CTC, cells were co-stained with 3′-(p-hydroxyphenyl) fluoroscein (HPF), a cell permeable fluorescent reporter that has widely been used to detect levels of highly reactive oxygen species like hydroxyl radicals (OH•) [13]. CTC staining of S. aureus grown in TSB-14 mM glucose revealed a healthy respiring sub-population (∼34%) at 24 h and a relatively smaller population of cells (13%) undergoing oxidative stress (Fig. 1B, Table S1). By 72 h the respiring population under these very same conditions increased to 79% (Fig. 1B, Table S1). This is expected as dependency on the TCA cycle and oxidative phosphorylation for cellular energetic needs increases in stationary phase, upon exhaustion of glucose from the media. Interestingly, S. aureus grown in TSB-35 mM glucose revealed an even larger population (∼61%) that reduced CTC at 24 h when compared to 14 mM glucose (Fig. 1B, Table S1). The increased reduction of CTC under these conditions could not have resulted from a corresponding increase in the rate of cellular respiration, as the ability of these cells to consume oxygen as a terminal electron acceptor had significantly decreased (Fig. 1C). These observations suggest that aerobic growth in excess glucose not only results in the inhibition of respiration, but may also promote the promiscuous transfer of electrons to alternate acceptors like CTC, due to a bottleneck in the ETC.
The transfer of electrons via a functional ETC has previously been proposed to ameliorate oxidative stress by curtailing the single electron reduction of oxygen to superoxide radicals (O2•−), a precursor of the highly reactive hydroxyl radical (OH•) [14]. Hence, we argued that the decreased functionality of ETC observed for cells grown in excess glucose may eventually promote the production of ROS. Although we did not observe OH• at 24 h of growth, we detected a dramatic increase of HPF stained cells by 72 h of growth in TSB-35 mM glucose but not in TSB-14 mM glucose (Fig. 1B, Table S1). The temporal production of ROS was confirmed by electron paramagnetic resonance (EPR) spectroscopic analysis of samples incubated with the spin probe, 1-hydroxy-methoxycarnonyl-2,2,5,5-tetramethyl-pyrrolidine hydrochloride (CM-H). Cells grown in TSB-35 mM glucose exhibited approximately 3-fold increase in EPR peak amplitude by 72 h relative to those grown in TSB-14 mM glucose (Fig. 1D). To determine the chemical nature and relative levels of various ROS produced, we incubated samples with either superoxide dismutase (SOD; O2•− scavenger) or dimethyl thiourea (DMTU; OH• scavenger) prior to the addition of CM-H. This approach revealed that cells undergoing cell death produced both superoxide and hydroxyl radicals (Fig. S1).
An abundance of cellular ROS mediates several types of DNA damage, including single and double stranded breaks that lead to DNA fragmentation [15]. We performed TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assays on S. aureus undergoing oxidative stress to estimate the population of cells undergoing DNA fragmentation by flow cytometry. Consistent with the temporal pattern of ROS production observed earlier, we detected a sub-population of cells with fragmented DNA (TUNEL positive) by 72 h of growth under excess glucose conditions (Fig. 1E). Notably, only minimal TUNEL staining (Fig. 1E) was detected after 72 h for cultures supplemented with 14 mM glucose. Collectively, these observations suggest that cell death resulting from growth under excess glucose exhibits multiple hallmarks of eukaryotic PCD. Interestingly, the phenotypic hallmarks of PCD were not restricted to growth of S. aureus under glucose rich conditions alone, but were also observed when grown in the presence of excess fructose, mannitol and sucrose suggesting a strong association between carbon catabolism and cell death (Fig. S2).
Acetate potentiates cell death under low pH
How does excess glucose mediate cell death? It is well known that S. aureus cultured in excess glucose undergoes CcpA-mediated catabolite repression [16]. This ensures that acetate accumulates in the media as a byproduct of glucose catabolism. However, once glucose is completely exhausted from the media, the TCA cycle is progressively relieved of CcpA repression and excreted acetate is oxidized to generate energy required for subsequent growth [16]. Growth of S. aureus in TSB-14 mM glucose displayed such a classic diauxie, where glucose was consumed within 5 h and subsequent growth was dependent on the consumption of acetate by 9 h (Fig. 2A–C). The temporal dynamics of acetate levels in the media were also reflected in the pH shift of the culture supernatant from 7.2 to 5.5 during acetate accumulation and from 5.5 to 7.2 during its depletion (Fig. 2C, 2D). As observed previously [17], growth of S. aureus in TSB-35 mM glucose did not display the expected diauxic shift (Fig. 2A). Although excess glucose was consumed within 9 h, acetate remained unutilized and the pH of the culture was maintained at 4.6 (Fig. 2B–D).

Based on these observations, we hypothesized that cells grown in excess glucose would eventually be inhibited by high concentrations of acetate and low pH. The growth inhibitory effects of weak organic acids like acetate are largely dependent on pH [18]. As the extracellular pH nears the pKa of acetate (pKa = 4.76), the protonated (uncharged) membrane permeant form of the acid (CH3COOH) replaces its corresponding ionic forms (CH3COO−; H+), thus allowing the former species to passively breach bacterial membranes and dissociate within their relatively neutral cytoplasm [18]. If left unchecked, such an event may lead to growth inhibition and lethality through cytoplasmic acidification [18]. Given that the pKa of acetate is easily met during growth in excess glucose, it seemed plausible that this acidic metabolite may represent a physiological trigger for cell death. To test whether acetate was capable of inhibiting S. aureus growth under acidic conditions, we buffered TSB at pH 4.8 using 30 mM HOMOPIPES (homopiperazine-N,N′-bis-2-(ethanesulfonic acid) and challenged cultures with the sodium salt of acetate. As expected, acetate inhibited the growth of S. aureus under these conditions, but neither acidic pH alone nor addition of an equimolar concentration of sodium chloride (50 mM) inhibited growth to the same extent as sodium acetate (Fig. 2E). Further, the addition of a non-metabolizable weak acid, benzoate (pKa = 4.2) was as toxic as acetate under low pH (Fig. 2E). These observations suggest that acetate mediated growth inhibition is a direct consequence of intracellular acidification and not due to secondary metabolites resulting from intracellular catabolism of acetate.
To confirm that cell death is dependent on the weak acid properties of acetate, we grew S. aureus in TSB-35 mM glucose that was buffered to a pH of 7.3 with 50 mM MOPS (3-(N-morpholino) propanesulfonic acid). We reasoned that although cells would utilize excess glucose to generate acetate, the relatively neutral pH of the medium would allow it to remain in the ionic state and prevent it from permeating and acidifying the interior of cells. Indeed as predicted, S. aureus under these conditions did not undergo cell death despite its growth in excess glucose (Fig. 2F). Remarkably, we also observed a dramatic reduction in the generation of ROS (Fig. 2G–H, Table S1), decreased DNA damage (Fig. 2I) and comparable rates of respiration relative to wild-type (Fig. 2J) under these conditions, suggesting that excess glucose per se was not responsible for the phenotypes associated with cell death. Rather these observations collectively demonstrate that acetate, a metabolic byproduct of glucose catabolism triggered these phenotypes under acidic pH.
CidR-dependent overflow metabolic pathways regulate staphylococcal cell death
Although acetate is primarily produced in S. aureus by the phosphotransacetylase (Pta)-acetate kinase (AckA) pathway, this metabolic pathway is unlikely to be directly involved in cell death as its activity is evident even during growth in 14 mM glucose, a condition where cell death is not triggered [19]. Additionally, disruption of this pathway surprisingly enhanced the rate of cell death during growth despite a decrease in acetate production [19]. This led us to reason that acetate-dependent cell death must be controlled by an alternate pathway, such as CidC. The cidC gene encodes a pyruvate oxidase that directly converts pyruvate to acetate and carbon dioxide and its expression is partly under the control of the regulator, CidR, whose activity is up-regulated in response to excess glucose [7], [19]. As the alsSD metabolic operon that results in the conversion of pyruvate to acetoin is also co-regulated by CidR, we hypothesized that both these pathways may modulate acetate-dependent cell death by competition for their common substrate, pyruvate, under conditions of excess glucose (Fig. 3A).

To test this hypothesis, we initially determined the levels of acetate and acetoin in 24 h culture supernatants of both ΔcidC and ΔalsSD mutants relative to WT. Compared to the wild-type strain, the ΔcidC mutant grown in TSB-35 mM glucose accumulated less acetate and relatively higher levels of acetoin (Fig. 3B, 3C). Conversely the ΔalsSD mutant excreted an excess of acetate (Fig. 3B) suggesting that both pathways competitively displaced pyruvate. We then tested the effects of cidC and alsSD pathways on cell death. In agreement with earlier studies [17], [20], mutation of either of these pathways resulted in opposing survival trends in stationary phase. Accordingly, a metabolic block in CidC activity (ΔcidC) enhanced stationary phase survival, while that of AlsSD (ΔalsSD) resulted in an increased rate of cell death compared to the wild-type strain (Fig. 3D). Consistent with the increased survival of the ΔcidC mutant and in contrast to WT and the ΔalsSD mutant, HPF-CTC double staining of 72 h cultures revealed a healthy population of respiring cells that exhibited low levels of ROS (Fig. 3E, Table S1), a phenotype that was also confirmed by EPR spectroscopy (Fig. 3F). In addition, flow cytometry detected fewer TUNEL-positive cells in the ΔcidC mutant, suggesting decreased DNA damage in these cells (Fig. 3G). Indeed, the cell death phenotypes associated with both ΔcidC and ΔalsSD mutants could be complemented in trans (Fig. S3) confirming their role in modulating cell death. Taken together, these data support the hypothesis that both CidC and AlsSD pathways modulate cell death by controlling flux through the pyruvate node.
Surprisingly, the ΔalsSD mutant generated ROS and exhibited DNA damage at levels similar to the wild-type strain (Fig. 3E–G, Table S1) despite an increase in loss of viability (Fig. 3D). This raised the possibility that in addition to affecting excreted acetate levels, there may be additional mechanisms by which the ΔalsSD mutant regulates cell death. To test this hypothesis we constructed a double ΔcidCΔalsSD mutant. We reasoned that if regulation of extracellular acetate by substrate competition was the primary mechanism by which AlsSD modulated cell death, then the ΔcidCΔalsSD double mutant would phenocopy the ΔcidC mutant. However, our results clearly demonstrate that the ΔcidCΔalsSD double mutant exhibited increased loss of viability (Fig. 3D), increased generation of ROS (Fig. 3E–F, Table S1) and excessive levels of DNA damage (Fig. 3G) than the ΔcidC mutant despite excreting comparable levels of acetate (Fig. 3B). Such an effect may occur if mutation of alsSD caused cells to be more susceptible to lower concentrations of weak acids in the culture media. Indeed, growth of ΔalsSD mutants challenged with acetate, lactate or pyruvate was more easily inhibited than wild-type (Fig. S4).
We tested two plausible hypotheses to explain the observed hyper-susceptibility of ΔalsSD mutants to weak acid stress. First, we argued that the end product of AlsSD catabolism, acetoin, itself may be necessary to withstand weak acid stress as it is known to contribute to the maintenance of cellular redox status upon being converted to butanediol or serve as a carbon source during stationary phase of growth. To test this hypothesis we subjected the ΔalsSD mutant to pyruvic acid stress and asked whether supplementation of excess acetoin in culture could rescue the pyruvate-mediated growth inhibition of this mutant. Our results demonstrate that acetoin could not restore pyruvate-mediated growth inhibition of the ΔalsSD mutant (Fig. S5). Furthermore, transformation of the ΔalsSD mutant with a plasmid bearing the alsS gene alone (in the absence of its cognate partner, alsD) under the control of its native promoter was able to rescue this mutant from pyruvic acid-mediated stress to growth rates comparable to the parental control, thus excluding any role for acetoin in promoting weak acid resistance (Fig. 4A).

We next hypothesized that AlsSD might play an active and crucial role in detoxifying intracellular acidification that accrues from the deprotonation of weak organic acids in the relatively neutral bacterial cytoplasm [21]. In such a scenario, intracellular protons would be consumed during multiple stages of decarboxylation, first of pyruvate into acetolactate catalyzed by AlsS, followed by that of the acetolactate intermediate into acetoin by AlsD, both leading to a gradual alkalization of the cytoplasm during weak acid stress. Direct evidence confirming a role for AlsSD in regulating intracellular pH was obtained by using cells loaded with the fluorescent pH probe 5 (and 6-)-carboxyfluorescein succinimidyl ester (cFSE). Resting cells that were suspended in potassium phosphate buffer (pH 4.5) maintained a slightly acidic interior (pHinternal of 5.9; Fig. 4B, top), resulting in a transmembrane pH gradient (ΔpH = pHinternal - pHexternal) of approximately 1.4 units. Addition of pyruvate under these conditions initiated a pH gradient (ΔpH) decay across the membrane that was exacerbated in the ΔalsSD and ΔcidC ΔalsSD backgrounds compared to either the parental or ΔcidC strains (Fig. 4B, bottom). Although the pH gradient decay recovered and stabilized over time, the rate and magnitude of the pHi recovery in different strains appeared to be dependent on the activity of AlsSD (Fig. 4B). In the presence of pyruvate, both the parental control and the ΔcidC mutant displayed comparable recovery rates of (23.83±1.9)×10−3 min−1 and (27.45±5.5)×10−3 min−1, respectively, and reached a pHi comparable to those of control cells (untreated resting cells) within 20 minutes (Fig. 4B, 4C). In contrast, the pHi of both the ΔalsSD and ΔcidCΔalsSD mutants had stabilized approximately 0.2–0.3 units below that of the pyruvate treated parental control and exhibited significantly decreased pHi recovery rates (P<0.05) of (11.66±2.1)×10−3 min−1 and (5.24±3.8)×10−3 min−1, respectively leading to incomplete recovery from acidic stress even after 60 minutes (Fig. 4B, 4C). These data demonstrate a role for the enzymatic activity of AlsSD in countering weak acid mediated intracellular acidification of the bacterial cytoplasm.
Acetate and ROS synergistically promote cell death
In eukaryotes, there is increasing evidence that ROS plays a key role in mediating PCD [22], [23]. Given that the physiological induction of ROS in S. aureus is dependent on the accrual of extracellular acetate, we next asked whether cell death is a direct consequence of acetic acid-mediated intracellular acidification or is an indirect result of oxidative stress. To this end we devised a strategy to determine the contribution of intracellular acidification on triggering cell death, independent of the ROS generated after 72 h of growth under aerobic conditions.
Wild-type S. aureus was aerobically grown for 24 h in TSB-35 mM glucose, followed by a sudden shift to anaerobic conditions. While this process ensured as much acidification as aerobically grown cultures, it conveniently eliminated ROS (due to the absence of oxygen). Although it is plausible that a sudden transition of cultures to anaerobic conditions may induce cell death independent of acidic stress, we controlled for this possibility by performing a similar experiment with wild-type S. aureus grown in TSB-35 mM glucose buffered to a pH of 7.3 with 50 mM MOPS. Cell viabilities monitored over a 120 h period showed only minimal loss of viability for neutrally buffered cultures, suggesting that cell death following anaerobic transition was dependent on culture acidification, similar to aerobically grown cells (Fig. 5A). Most importantly, unbuffered cultures that were shifted to anaerobic conditions displayed a partial restoration of viability compared to their corresponding aerobic cultures (Fig. 5A). Similarly a partial rescue was also observed when well-aerated cultures grown for 24 h were left standing without further agitation to minimize aeration (Fig. S6). Together, these data suggest a contributory role for oxidative stress in cell death.

Consistent with trends in cell viability, HPF-CTC staining of 72 h unbuffered cultures confirmed the absence of ROS following transition to anaerobiosis and the presence of a respiring population in contrast to that observed for the same time period under aerobic conditions (compare Fig. 5B with Fig. 1B, Table S1). Surprisingly, there was also a dramatic reduction of TUNEL positive cells following the shift to anaerobiosis (Fig. 5C), suggesting that ROS rather than intracellular acidification played a crucial role in DNA damage.
Staphylococcal cell death affects biofilm development and pathogenesis
Cell death in staphylococcal biofilms is often spatially restricted to developing microcolonies [24]. Given that the CidR regulon is also actively expressed in microcolonies [24], we predicted that cell death may be modulated by both CidC and AlsSD activities and further contribute to the structural and developmental integrity of the maturing biofilm. To test these hypotheses, we assayed the ability of the ΔcidC and ΔalsSD mutants to form biofilms, relative to the wild-type strain on glass surfaces exposed to a continuous flow of nutrients. Wild type biofilms appeared as a confluent biomass frequently interspersed with microcolonies that differentiated from the primary biofilm mat. As previously noted, live/dead cell staining of wild-type biofilms confirmed that dead cell populations were predominantly localized within microcolonies (Fig. 6A). However compared to the wild-type strain, COMSTAT analysis of ΔcidC biofilms revealed significantly decreased total biofilm biomass and thickness, indicative of developmental defects during biofilm formation (Fig. 6B, 6C). Additionally, the roughness coefficients (a measure of the biofilm architectural heterogeneity) of the ΔcidC mutant biofilms were significantly lower than those of wild-type (Fig. 6D), a phenotype that was also consistent with the decreased ability of the ΔcidC mutant to differentiate into microcolonies. Finally, the ΔcidC biofilm revealed significantly less dead cell biomass compared to the parental strain, strongly suggestive of its involvement in promoting cell death in biofilms (Fig. S7). Similar to the ΔcidC biofilm, COMSTAT analysis of biofilms formed by the ΔalsSD mutant also exhibited decreased biofilm biomass and thickness compared to the wild-type strain (Fig. 6B, 6C). However it is unlikely that the observed decrease in biomass of one-day old ΔalsSD biofilms was due to an early developmental defect as they were able to differentiate into microcolonies and attain similar roughness coefficients to that of its isogenic wild-type strain (Fig. 6D). Rather, these defects are consistent with increased sensitivity of the AlsSD mutant to weak acids and low pH environments of biofilm microcolonies. Collectively, these observations suggest that the activities of both CidC and AlsSD regulate cell death at the population level to achieve optimal biomass and structural integrity during biofilm development.
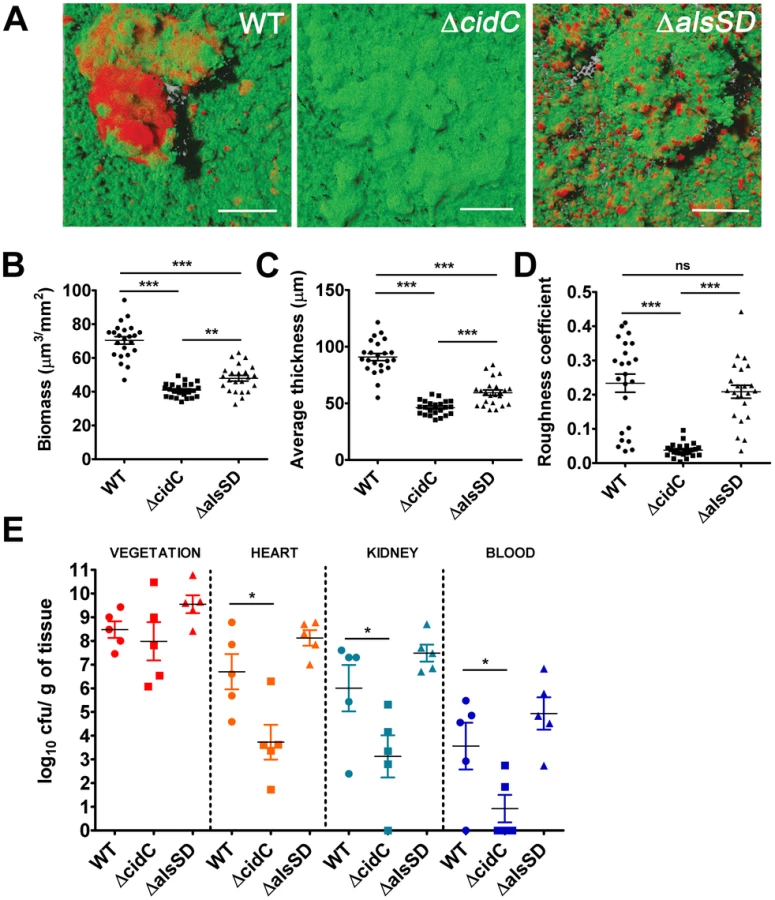
Since the ability of bacteria to develop as biofilms on heart valves constitutes the root cause of infective endocarditis, we speculated that staphylococcal cell death may also contribute towards pathogenesis in a rabbit model of infective endocarditis. This model not only provides an estimate of the organism's biofilm forming capability in vivo but also allows for the simultaneous assessment of embolization (dissemination of the bacterial vegetation to secondary sites due to blood flow associated shear forces in the heart). To test this hypothesis we induced left-sided endocarditis in rabbits and infected them with wild-type, ΔcidC and ΔalsSD mutant strains. At 48 h post-infection, the bacterial burden in the primary vegetation (heart valves), heart tissue, kidney and blood were determined. Consistent with being the primary site of biofilm infection, the heart valves exhibited the maximum bacterial burden (∼108 cfu/gm of tissue) among the various tissues harvested (Fig. 6E). Relative to the wild-type strain, both ΔcidC and ΔalsSD mutants had similar bacterial loads at this site (Fig. 6E) suggesting that these metabolic pathways did not affect the growth of these strains or their ability to colonize the primary infection sites in vivo. Interestingly however, the ΔcidC mutant displayed significantly decreased bacterial burdens in the blood and other secondary sites of infection including heart tissue (excluding valves) and kidneys (Fig. 6E). As bacterial colonization of these secondary sites primarily results from infectious emboli originating from the heart valve, it may be argued that the cell-death associated CidC pathway may play a role in metastasis of the valvular vegetation.
Discussion
In the present study, we demonstrate that both CidC and AlsSD pathways, that have traditionally been considered metabolic routes for excess carbon flow, antithetically modulate staphylococcal cell death by regulating the levels of excreted acetic acid. Exercising such metabolic control over cell death affords S. aureus a means to modulate biofilm development and possibly disperse and colonize alternate sites during the course of biofilm-associated infections.
How does acetate potentiate cell death? Our results reveal that both intracellular acidification and ROS generation may play a role in acetate dependent cell death. Although intracellular acidification can result from other fermentative metabolites like D - or L-lactate, we were unable to detect excretion of L-lactate and observed only small differences in the excretion of D-lactate in the ΔcidC and ΔalsSD mutants relative to WT (Fig. S8). Furthermore, given that the pKa of D-lactate (pKa = 3.86) is lower than acetate and its levels in culture supernatants were minute (∼35 fold less than acetate) we reasoned that it is unlikely to have a similar effect to that of acetic acid on cell death during aerobic growth. At the molecular level, it is not clear how acetate initiates ROS production. The evidence presented in this study suggests that acetate may contribute to a bottle-neck in electron transport by reducing the functionality of the respiratory chain. This could catalyze the promiscuous reduction of molecular oxygen and result in the production of ROS. Consistent with this argument, we have confirmed increased levels of superoxide and hydroxyl radicals following growth of S. aureus in 35 mM glucose, a condition that leads to acetate stress. Given that the pKa of superoxide anion is 4.88, it is very likely that this species freely traverses the cytoplasmic membrane and mediates oxidative damage during acetic acid stress. In addition to ROS, cytoplasmic acidification due to acetate influx itself can be a significant cause of cell death. Evidence for this conclusion arises from the observation that both metabolizable (acetate, lactate and pyruvate) and nonmetabolizable (benzoate) weak acids inhibit S. aureus growth. Such inhibition can result from acid catalyzed intracellular protein unfolding and aggregation. In support of this conclusion, a recent transcriptomic analysis of S. aureus challenged with the weak acid, lactate, revealed various clp genes (including clpB, clpC and clpP) involved in protein folding and recycling to be strongly up regulated [25].
Extending these findings to biofilms, we argue that the acidic pH microenvironments of biofilm microcolonies [26], [27] may spatially bias these biological structures as sites of respiratory inhibition and cell death. Weak acid metabolic byproducts like acetate and lactate are thought to accumulate within the biofilm interior, primarily due to diffusion limits resulting from reduced fluid flow and accumulation of biofilm matrix components [28]. We propose a model of staphylococcal PCD wherein the acidic pH within microcolonies activates expression of the CidR regulon [29] in a subpopulation of cells. This subsequently would lead to a feed-forward loop in which toxic acetate levels are reached through CidC activity. Ultimately cell death would ensue when macromolecular repair mechanisms are exhausted and cells within the biofilm are overwhelmed by the damaging effects of acetate (Fig. S9). To prevent a disproportionate number of cells from undergoing cell death, S. aureus co-expresses the AlsSD pathway along with CidC. Enzymatic activity of acetolactate synthase (AlsS) results in the condensation of two molecules of pyruvate to acetolactate and thereafter to acetoin by acetolactatae decarboxylase (AlsD). This effectively minimizes the carbon diverted to the generation of toxic acetate through the CidC pathway. More importantly evidence presented here also shows that the AlsSD pathway consumes protons from the cytoplasm and helps maintain pH homeostasis similar to the enterococci and lactobacilli [21], [30]. We contend that the ability of AlsSD to antagonize CidC activity effectively results in the modulation of intracellular pH and constitutes a robust mechanism to limit cell death and optimize biomass within the microenvironment of a microcolony.
The pathways that generate acetate vary among organisms. Whereas most bacteria, including E. coli and S. aureus generate acetate under aerobic conditions through the Pta-AckA and the CidC pathways, most eukaryotes (yeasts and mammals) lack these enzymes. Instead yeasts produce acetate as a natural byproduct of ethanol fermentation from acetaldehyde using acetaldehyde dehydrogenase [31]. Mammalian cells rarely generate significant quantities of acetate. But under certain conditions, acetate is produced by the enzymatic hydrolysis of acetyl-CoA in the cytoplasm [32]. Irrespective of the diversity in production routes, multiple studies have demonstrated that acetate itself can act as a potent inducer of PCD in yeasts and mammalian cells [33], [34]. Consistent with these studies, we demonstrate multiple hallmarks of eukaryotic PCD including respiratory dysfunction, generation of ROS and DNA fragmentation to be conserved during acetate mediated cell death in S. aureus. Further, similar to eukaryotic PCD [1], acetate-mediated cell death functions in a developmental context and appears to be crucial for optimal staphylococcal biofilm development.
It is noteworthy that two different activities of pyruvate oxidase (CidC, also annotated as PoxB in E. coli and SpxB in S. pneumoniae) have been described previously [35], [36]. In E. coli and S. aureus, this enzyme catalyses the decarboxylation of pyruvate into acetic acid and carbon dioxide, whereas acetyl phosphate and hydrogen peroxide are the predominant products of a similar reaction in L. plantarum and S. pneumoniae. Remarkably, both these reactions appear to induce stationary phase cell death in bacteria with either acetate or hydrogen peroxide as principal determinants of cell death [37]. Additionally, similar to S. aureus, cell death due to pyruvate oxidase activity is associated with apoptotic hallmarks in S. pneumoniae [37]. These observations appear to clearly mark pyruvate oxidase activity as a suicidal marker in bacteria.
Finally, what are the biological implications of regulating PCD? We used a well-established rabbit model of infective endocarditis to assess the effects of altering PCD on in vivo biofilm development. S. aureus injected intravenously in rabbits is rapidly cleared from the blood within the first 30 minutes leaving only minute residual amounts to linger over longer periods of time [38]. However any injury to the heart valves marks a preferred site for bacterial colonization and eventual development into a biofilm (vegetation). The pathological progression of infective endocarditis subsequently involves embolization of bacterial vegetations to alternate sites including surrounding heart tissues and other peripheral organs like the brain and kidneys [39]. This process not only poses a constant seeding source of infection but also hinders the normal functioning of peripheral organs and is often associated with a high degree of mortality [39]. Our investigations failed to reveal a colonization defect of the ΔcidC and ΔalsSD mutants on heart valves relative to the wild-type strain. However we observed a significant decrease in ΔcidC burdens in the blood, heart and kidneys after 48 h of infection. Although not conclusive, these findings strongly suggest that the ΔcidC mutant had lower rates of dissemination to secondary infection sites in vivo. Alternately, it is also possible that the ΔcidC mutant exhibits tissue specific fitness and survival defects in vivo. Either way, these findings suggest that alterations to the activity of cell-death associated metabolic pathways during biofilm development could affect staphylococcal pathogenesis.
In conclusion, the activation of pathways that generate metabolic acids from glucose during carbon-overflow and oxygen replete conditions have long been considered paradoxical in bacteria that are capable of undergoing oxidative phosphorylation, as it results in low energy yields, potentially toxic acid by-products and activation of cell death pathways [40]. Based on the current study we propose that the extracellular accumulation of metabolic acids is a developmental strategy that bacteria undertake to initiate cell death, a necessary precursor to optimal biofilm development. The initiation of staphylococcal cell death by intracellular acidification bears some striking resemblance to eukaryotic PCD. For instance, the dimerization and insertion of the pro-apoptotic modulator, Bax, into the membrane is thought to be triggered by intracellular acidification of eukaryotic cells [41] just prior to the release of cytochrome c into the cytoplasm. In this regard, it is possible that membrane oligomerization of CidAB and LrgAB (functional analogs of Bax and Bcl-2 in S. aureus, respectively) may also be initiated following intracellular acidification. Additionally, cytoplasmic acidification in eukaryotes also activates caspases, essential components of the apoptotic pathway [42]. Collectively, these observations are suggestive of a conserved role for glycolysis-mediated intracellular pH regulation in the modulation of PCD in eukaryotes and prokaryotes.
Materials and Methods
Ethics
Animal experiments were conducted in compliance with a protocol (# 12-048-08-FC) approved by the Institutional Animal Care and Use Committee (IACUC). The University of Nebraska Medical Center is accredited by the Association of for Assessment and Accreditation of Laboratory Animal Care International (AALAC). In addition, all animals at the University of Nebraska Medical Center are maintained in accordance with the Animal Welfare Act and the DHHS “Guide for the Care and Use of Laboratory Animals.”
Bacterial strains, plasmids and growth conditions
Strains and plasmids used in this study are listed in Table S2. The ΔcidCΔalsSD double mutant was created by moving the ΔcidC::erm allele from KB1058 into the ΔalsSD mutant (UAMS-1489) using bacteriophage Φ11-mediated transduction. In addition to growth on selective antibiotic media, the ΔcidC transductants were confirmed phenotypically and by PCR using the following primer pairs: cidC UP (5′-CACATGCATTTGGCACAGCT-3′) and cidC DN (5′-TGCTCATGCCTGCATTACCA-3′). The plasmid, pVCT2, containing the alsS gene with its native promoter was constructed by amplifying an approximately 2-kb region from the UAMS-1 genome using the primers, alsS-comp-F (5′-GATCGAGCTCTCCCTTATAATCACTCCCTTCA-3′) and alsS-comp-R (5′-AGTCTCTAGATGTGCCTAATGTACCATGTTG-3′), and inserting the resulting DNA fragment into the Sac1 and Xba1 sites of the shuttle vector, pLI50 [43]. Similarly, plasmid pVCT3 (containing cidC gene with its native promoter) was amplified from a ΔcidAB double mutant using primers, cidC comp-F (5′-GATCGAATTCACTCATTATTTGTGATTCCTCA-3′) and cidC comp-R (5′-AGTCGTCGACCAATTCAGTACAATCATTTGTG-3′). The resulting amplification product was inserted into the EcoR1 and Sal1 sites of pLI50. Subsequently, both pVCT2 and pVCT3 were transformed into RN4220 and transduced into the ΔalsSD and ΔcidC mutants respectively, using bacteriophage ϕ11 for phenotypic complementation.
E. coli cultures were grown in Luria Bertani (LB) broth. S. aureus cultures were grown in trypticase soy broth (TSB) supplemented with 35 mM glucose (unless specified otherwise). Bacterial cultures were aerobically grown at 37°C in either Erlenmeyer flasks fitted with bug stoppers to minimize evaporation during long-term growth or in 96-well flat, clear bottom micro-titer plates. For anaerobic growth, cultures were supplemented with cysteine (0.5 mg/ml) and 10 mM nitrate (or fumarate) and agitated at 250 rpm in an anaerobic hut. When necessary, antibiotics were added to cultures as follows: ampicillin (100 µg/ml); erythromycin (5 µg/ml); tetracycline (10 µg/ml); and chloramphenicol (10 µg/ml).
Flow cytometry
All analyses were performed using 1 - and 3 - day old stationary phase cultures of S. aureus on a BD LSRII flow cytometer (Beckton and Dickinson, San Jose, California). Cell samples were washed twice and diluted to a final concentration of 107 cells per ml in PBS. Cells were stained for 30 min with 5-cyano-2,3-ditolyl tetrazolium chloride (CTC, 5 mM) and 3-(p-hydroxyphenyl) fluorescein (HPF, 15 µM) followed by FACS analyses at a flow rate of ∼1000 cells per second. A total of 10000 events were collected for each sample. Bacteria were discriminated from background using a combination of forward scattered ligt (FSC) and side scattered light (SSC). Samples were excited at 488 nm using an argon laser and HPF emission was detected at 530±30 nm (with a 505 nm long-pass mirror) whereas CTC emission was detected at 695±40 nm (with a 685 nm long-pass mirror). Raw data were analyzed using the FlowJo software.
TUNEL assay
Quantitative assessment of DNA fragmentation was performed using the ApoDirect kit (BD bioscience). Samples were collected at the appropriate time points and fixed in 1% paraformaldehyde for 30 minutes. Cells were then washed twice in PBS, resuspended in 70% ethanol and stored at −20°C. Subsequent labeling of 3-OH ends of fragmented DNA was performed according to the manufacturer's instructions. Flow cytometry to detect TUNEL positive cells was performed as previously described [4].
Electron paramagnetic resonance (EPR) spectroscopy
Aliquots from stationary phase cultures (1 - and 3 days) were withdrawn and resuspended to an OD600 of 10 units in 1 ml KDD buffer (Krebs-HEPES buffer, pH 7.4; 99 mM NaCl, 4.69 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 1.03 mM KH2PO4, 5.6 mM D-glucose, 20 mM HEPES, 5 µM DETC and 25 µM deferoxamine). The resuspended culture aliquots were then incubated with 200 µM of cell-permeable ROS sensitive spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH; Noxygen Science Transfer and Diagnostics, Elzach, Germany) for 15 minutes at room temperature prior to analysis using a Bruker e-scan EPR spectrometer with the following settings: field sweep width, 60.0 gauss; microwave frequency, 9.75 kHz; microwave power, 21.90 mW; modulation amplitude, 2.37 gauss; conversion time, 10.24 ms; time constant, 40.96 ms. To identify the nature of ROS produced, cells resuspended in KDD buffer were incubated with either 400 units of superoxide dismutase (SOD; O2•− scavenger) or cell permeable dimethyl thiourea (20 mM DMTU; OH• scavenger) prior to the addition of CMH.
Oxygen consumption
S. aureus was cultured at 37°C in TSB supplemented with either 14 or 35 mM glucose and aerated at 250 rpm with a flask-to-medium ratio of 10∶1 for 24 h. Cultures were subsequently diluted to an OD600 of 0.1 in fresh TSB (14 mM glucose). Oxygen consumption rates were determined for a period of 30 minutes at 37°C by using a MitoXpress oxygen-sensitive probe (Luxcel Biosciences) according to the manufacturer's instructions. The data were normalized to the corresponding OD600 units.
Metabolite analyses
For these analyses, bacterial growth was allowed to proceed at 37°C and 200 rpm in BugStopper-sealed flasks containing TSB (35 mM glucose) in a 1∶10, flask to volume ratio. Preliminary experiments suggested that the assayed metabolite by-products were not significantly consumed following exhaustion of glucose from the media for up to 24 h. Therefore metabolite excretion profiles were determined from culture supernatants that were harvested at 24 h of growth. Acetate and glucose from culture supernatants were measured using commercial kits (R-Biopharm, Marshall, MI), according to the manufacturer's instructions. Acetoin was measured as previously described [44].
Measurement of bacterial growth
Overnight grown (16 to 18-hr) S. aureus cultures were resuspended to an OD600 of 0.06 in TSB (35 mM glucose). Bacterial suspensions were dispensed into 96-well microtiter plates and grown for 24 h at 37°C in a Tecan infinite 200 spectrophotometer under maximum aeration. The absorbance signals (OD600) were recorded every 30 minutes for the entire period of growth. For various experiments bacteria were challenged with the following compounds (final concentrations): acetic acid (30 mM), lactic acid (40 mM), pyruvic acid (30 mM) and acetoin (10 mM).
Intracellular pH determination
Intracellular pH was determined as previously described [30] with the minor modifications. Briefly, bacterial cells were grown in TSB (35 mM glucose) and 1 mL was harvested by centrifugation upon reaching an OD600 of 2. Cells were washed twice with an equal volume of 10 mM potassium phosphate buffer (pH 7) and resuspended in an equal volume of the same buffer. To load cells with the intracellular pH probe, 10 µl of 1 mM CFDA SE (5-(and 6) - carboxyfluorescein diacetate succinimidyl ester) was added to the suspension and incubated for 15 minutes at 30°C. Excess dye was removed by incubating the cells for 15 minutes at 30°C in potassium phosphate buffer (pH 7) containing 10 mM glucose. Cells were subsequently washed twice in the same buffer and finally resuspended in 50 mM potassium phosphate buffer (pH 4.5). Labeled cells were kept on ice until use.
To measure intracellular pH, 100 µl of the labeled cell suspensions were introduced into a 96-well flat bottom, black polystyrene plate (COSTAR 3916). Fluorescence was measured using a Tecan Infinite 200 spectrofluorimeter with excitation and emission wavelength set at 490 nm and 525 nm, respectively. Fluorescence emission units were converted to pH units using a standard calibration curve derived from labeled cells whose internal pH was equilibrated to the external pH (in citric acid buffers) ranging from pH 4 to 8, by the addition of 1 mM valinomycin and 1 mM nigericin.
Biofilm assays and confocal microscopy
Biofilms were grown in either FC280 or FC285 flow-cell systems (Biosurfaces Technology Inc, Bozeman, MT) and were analyzed by CLSM as described previously [45]. Briefly, biofilms stained with SYTO-9 (1.3 µM final concentration) and TOTO-3 (2 µM final concentration) fluorophores were excited with the 488 nm and 633 nm lasers respectively, and the emissions were collected using a 525±25 nm and 680±30 nm band-pass filter. For pictorial representation, the biofilms were imaged using an Achroplan 40×0.8 n.a. water dipping objective and for COMSTAT image analysis, images were acquired using a 20×1.2 n.a. dry objective to achieve a larger biofilm surface area for statistical purposes. Regions of interest within the biofilms were selected from similar areas within each flow-cell chamber and each confocal experiment was repeated a minimum of three times. Biofilm architecture was characterized using the COMSTAT software and measures of total biomass, average thickness, maximum height and roughness coefficients were determined [46]. Images were rendered using Imaris software (Bitplane, Saint Paul, MN).
Experimental endocarditis
Experimental endocarditis on the aortic valve of female New Zealand White rabbits (3 kg) were carried out as previously described with minor modifications [47]. Briefly, rabbits were anesthetized by intramuscular injection of ketamine hydrochloride (35–50 mg/kg), xylazine (2.5–6 mg/kg) and atropine (0.005–0.01 mg/kg) cocktail. An incision was made dextrolateral to the trachea and a polyethylene catheter (Becton Dickinson, MD) was then introduced into the left ventricle via the right carotid artery to produce sterile thrombotic endocarditis, and the skin incision sutured. To induce bacterial endocarditis, animals were intravenously challenged with 1 ml inocula (105 cfu/ml) via the marginal ear vein 24 h after catheterization. The animals were challenged with either the wild-type or mutant strains (ΔcidC and ΔalsSD) and subsequently (48 h post-infection) euthanized by lethal injection of a solution containing sodium pentobarbital (200 mg/kg). Bacterial loads from various tissue homogenates were determined by serial dilutions on THB agar plates.
Supporting Information
Zdroje
1. Ameisen JC (2001) The origin and evolution of programmed cell death. eLS: Wiley.
2. BaylesKW (2007) The biological role of death and lysis in biofilm development. Nat Rev Microbiol 5 : 721–726.
3. RiceKC, BaylesKW (2008) Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev 72 : 85–109 table of contents
4. DwyerDJ, CamachoDM, KohanskiMA, CalluraJM, CollinsJJ (2012) Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis. Mol Cell 46 : 561–572.
5. BosJ, YakhninaAA, GitaiZ (2012) BapE DNA endonuclease induces an apoptotic-like response to DNA damage in Caulobacter. Proc Natl Acad Sci U S A 109 : 18096–18101.
6. ErentalA, SharonI, Engelberg-KulkaH (2012) Two programmed cell death systems in Escherichia coli: an apoptotic-like death is inhibited by the mazEF-mediated death pathway. PLoS Biol 10: e1001281.
7. YangSJ, DunmanPM, ProjanSJ, BaylesKW (2006) Characterization of the Staphylococcus aureus CidR regulon: elucidation of a novel role for acetoin metabolism in cell death and lysis. Mol Microbiol 60 : 458–468.
8. SonensheinAL (2007) Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol 5 : 917–927.
9. NahkuR, ValgepeaK, LahtveePJ, ErmS, AbnerK, et al. (2010) Specific growth rate dependent transcriptome profiling of Escherichia coli K12 MG1655 in accelerostat cultures. J Biotechnol 145 : 60–65.
10. SadykovMR, BaylesKW (2012) The control of death and lysis in staphylococcal biofilms: a coordination of physiological signals. Curr Opin Microbiol 15 : 211–215.
11. AndersenJL, KornbluthS (2013) The tangled circuitry of metabolism and apoptosis. Mol Cell 49 : 399–410.
12. SmithJJ, McFetersGA (1997) Mechanisms of INT (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl tetrazolium chloride), and CTC (5-cyano-2,3-ditolyl tetrazolium chloride) reduction in Escherichia coli K-12. Journal of Microbiological Methods 29 : 161–175.
13. SetsukinaiK, UranoY, KakinumaK, MajimaHJ, NaganoT (2003) Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 278 : 3170–3175.
14. RezaikiL, CesselinB, YamamotoY, VidoK, van WestE, et al. (2004) Respiration metabolism reduces oxidative and acid stress to improve long-term survival of Lactococcus lactis. Mol Microbiol 53 : 1331–1342.
15. HiguchiY (2003) Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem Pharmacol 66 : 1527–1535.
16. SomervilleGA, ProctorRA (2009) At the crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol Mol Biol Rev 73 : 233–248.
17. PattonTG, RiceKC, FosterMK, BaylesKW (2005) The Staphylococcus aureus cidC gene encodes a pyruvate oxidase that affects acetate metabolism and cell death in stationary phase. Mol Microbiol 56 : 1664–1674.
18. Booth IR, Stratford M (2003) Acidulants and low pH. In: Russell NJ, Gould GW, editors. Food preservatives. 2nd ed. New York: Kluwer Academic/Plenum Publishers. 25–42 p.
19. SadykovMR, ThomasVC, MarshallDD, WenstromCJ, MoormeierDE, et al. (2013) Inactivation of the Pta-AckA pathway causes cell death in Staphylococcus aureus. J Bacteriol 195 : 3035–3044.
20. TsangLH, CassatJE, ShawLN, BeenkenKE, SmeltzerMS (2008) Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PLoS One 3: e3361.
21. TsauJL, GuffantiAA, MontvilleTJ (1992) Conversion of Pyruvate to Acetoin Helps To Maintain pH Homeostasis in Lactobacillus plantarum. Appl Environ Microbiol 58 : 891–894.
22. SimonHU, Haj-YehiaA, Levi-SchafferF (2000) Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5 : 415–418.
23. GranotD, LevineA, Dor-HefetzE (2003) Sugar-induced apoptosis in yeast cells. FEMS Yeast Res 4 : 7–13.
24. MoormeierDE, EndresJL, MannEE, SadykovMR, HorswillAR, et al. (2013) Use of microfluidic technology to analyze gene expression during Staphylococcus aureus biofilm formation reveals distinct physiological niches. Appl Environ Microbiol 79 : 3413–3424.
25. RodeTM, MoretroT, LangsrudS, LangsrudO, VogtG, et al. (2010) Responses of Staphylococcus aureus exposed to HCl and organic acid stress. Can J Microbiol 56 : 777–792.
26. HidalgoG, BurnsA, HerzE, HayAG, HoustonPL, et al. (2009) Functional tomographic fluorescence imaging of pH microenvironments in microbial biofilms by use of silica nanoparticle sensors. Appl Environ Microbiol 75 : 7426–7435.
27. HunterRC, BeveridgeTJ (2005) Application of a pH-sensitive fluoroprobe (C-SNARF-4) for pH microenvironment analysis in Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71 : 2501–2510.
28. StewartPS (2003) Diffusion in biofilms. J Bacteriol 185 : 1485–1491.
29. RiceKC, NelsonJB, PattonTG, YangSJ, BaylesKW (2005) Acetic acid induces expression of the Staphylococcus aureus cidABC and lrgAB murein hydrolase regulator operons. J Bacteriol 187 : 813–821.
30. RepizoGD, MorteraP, MagniC (2011) Disruption of the alsSD operon of Enterococcus faecalis impairs growth on pyruvate at low pH. Microbiology 157 : 2708–2719.
31. WellenKE, ThompsonCB (2012) A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 13 : 270–276.
32. WolfeAJ (2005) The acetate switch. Microbiol Mol Biol Rev 69 : 12–50.
33. LudovicoP, SousaMJ, SilvaMT, LeaoC, Corte-RealM (2001) Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 147 : 2409–2415.
34. MarquesC, OliveiraCS, AlvesS, ChavesSR, CoutinhoOP, et al. (2013) Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis 4: e507.
35. TittmannK, GolbikR, GhislaS, HubnerG (2000) Mechanism of elementary catalytic steps of pyruvate oxidase from Lactobacillus plantarum. Biochemistry 39 : 10747–10754.
36. RussellP, HagerLP, GennisRB (1977) Characterization of the proteolytic activation of pyruvate oxidase. Control by specific ligands and by the flavin oxidation-reduction state. J Biol Chem 252 : 7877–7882.
37. Regev-YochayG, TrzcinskiK, ThompsonCM, LipsitchM, MalleyR (2007) SpxB is a suicide gene of Streptococcus pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J Bacteriol 189 : 6532–6539.
38. DhawanVK, YeamanMR, CheungAL, KimE, SullamPM, et al. (1997) Phenotypic resistance to thrombin-induced platelet microbicidal protein in vitro is correlated with enhanced virulence in experimental endocarditis due to Staphylococcus aureus. Infect Immun 65 : 3293–3299.
39. Herzberg MC (2000) Persistence of infective endocarditis. In: Nataro JP, Blaser, M. J., Cunningham-Rundles, S., editor. Persistent bacterial infections Washington, DC: ASM Press. pp. 470.
40. ZhuangK, VemuriGN, MahadevanR (2011) Economics of membrane occupancy and respiro-fermentation. Mol Syst Biol 7 : 500.
41. XieZ, SchendelS, MatsuyamaS, ReedJC (1998) Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry 37 : 6410–6418.
42. FurlongIJ, AscasoR, Lopez RivasA, CollinsMK (1997) Intracellular acidification induces apoptosis by stimulating ICE-like protease activity. J Cell Sci 110(Pt 5): 653–661.
43. LeeCY, BuranenSL, YeZH (1991) Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103 : 101–105.
44. NicholsonWL (2008) The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanediol dehydrogenase. Appl Environ Microbiol 74 : 6832–6838.
45. MannEE, RiceKC, BolesBR, EndresJL, RanjitD, et al. (2009) Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4: e5822.
46. HeydornA, NielsenAT, HentzerM, SternbergC, GivskovM, et al. (2000) Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146 : 2395–2407.
47. ThurlowLR, ThomasVC, NarayananS, OlsonS, FlemingSD, et al. (2010) Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect Immun 78 : 4936–4943.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability