
Progressive familial intrahepatic cholestasis type 2 – paediatric patients followed at the Paediatric Clinic of the 2nd Medical Faculty, University Hospital Motol, Prague
Progressive familial intrahepatic cholestasis type 2 – paediatric patients followed at the Paediatric Clinic of the 2nd Medical Faculty, University Hospital Motol, Prague
Summary:
Progressive familial intrahepatic cholestasis type 2 is an autosomal recessive cholestatic liver disease caused by a deficiency in canalicular ATP-dependent bile salt export pump BSEP. We present case reports and collected data of six Czech patients suffering from this disease, diagnosed between 2005 and 2015, in whom seven new mutations in ABCB11 were revealed by molecular analysis.
Key words:
progressive familial intrahepatic cholestasis type 2 - BSEP – ursodeoxycholic acid – pruritus – liver transplantation
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
Submitted:
20. 11. 2015
Accepted:
27. 11. 2015
Autoři:
R. Kotalová 1; E. Sticová 2; M. Jirsa 3
Působiště autorů:
Pediatrická klinika 2. LF UK a FN v Motole, Praha
1; Pracoviště klinické a transplantační patologie, Transplantcentrum, IKEM, Praha
2; Laboratoř experimentální hepatologie, Centrum experimentální medicíny, IKEM, Praha
3
Vyšlo v časopise:
Gastroent Hepatol 2015; 69(6): 547-553
Kategorie:
Dětská gastroenterologie a hepatologie: původní práce
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amgh2015547
Souhrn
Progresivní familiární intrahepatální cholestáza 2. typu je autozomálně recesivně dědičná cholestáza podmíněná defektem ATP dependentní pumpy žlučových kyselin BSEP. V práci jsou uvedeny kazuistiky a souborná data šesti dětí s tímto onemocněním diagnostikovaných v ČR v letech 2005 – 2015, vč. výsledků jejich molekulárně genetických analýz, kterými bylo nalezeno sedm nových mutací v genu ABCB11.
Klíčová slova:
progresivní familiární intahepatální cholestáza 2. typu – BSEP – ursodeoxycholová kyselina – pruritus – transplantace jater
Úvod
Progresivní familiární intrahepatální cholestáza (PFIC) představuje skupinu autozomálně recesivně dědičných cholestáz. Jsou popsány čtyři typy onemocnění PFIC 1 – 4. Jejich incidence se předpokládá 1 : 50 000 – 100 000 živě narozených, přesná prevalence onemocnění není známa. PFIC je příčinou 10 – 15 % případů neonatální cholestázy (NCH) a tvoří 10 – 15 % indikací transplantací jater (TJ) v dětském věku [1].
PFIC2 (OMIM #601847) je způsobena defektem v genu ABCB11 kódujícím ATP dependentní pumpu žlučových kyselin konjugovaných s taurinem či s glycinem nazývanou BSEP (bile salt export pump) [2]. Gen ABCB11 se nachází na dlouhém raménku druhého chromozomu (2q24) a sestává z 28 exonů. Protein BSEP je exprimován výlučně v kanalikulární membráně hepatocytů [3,4].
Významnou úlohu v regulaci transkripce genu ABCB11 hraje nukleární transkripční faktor farnesoid X-aktivovaný receptor (FXR), který je zároveň důležitým regulátorem homeostázy cholesterolu [5]. FXR po navázání žlučové kyseliny (ŽK) jakožto nízkomolekulárního ligandu vytváří heterodimer s retinoid X-receptorem (RXR) vázajícím kyselinu retinovou. Takto vzniklý transkripční komplex aktivuje transkripci genu ABCB11. K aktivaci dochází při zvýšené koncentraci ŽK v cytoplazmě hepatocytů, a to buď vychytávaných z portální krve, nebo syntetizovaných de novo. Zpětnovazebná regulace transkripce zprostředkovaná FXR/ RXR představuje významný ochranný mechanizmus hepatocytů proti toxicitě žlučových kyselin při cholestáze.
Klinicky se PFIC2 obvykle manifestuje v prvních měsících života cholestatickou žloutenkou, těžkým pruritem, hepatosplenomegalií, retardací růstu a psychického vývoje. Bývají přítomny symptomy deficitu vitaminů rozpustných v tucích: koagulopatie, osteopenie a neuromuskulární poruchy [6], mohou vznikat žlučové konkrementy. Bez adekvátní léčby choroba progreduje do jaterní fibrózy až cirhózy a končí obvykle smrtí v důsledku jaterního selhání v první či vzácněji ve druhé dekádě.
Konzervativní léčba zahrnuje podávání kyseliny ursodeoxycholové (UDCA), cholestyraminu a rifampicinu a bývá obvykle bez většího efektu, potřebná je i substituce vitaminů rozpustných v tucích. Chirurgická léčba – ileální bypass nebo různé typy parciální externí biliární diverze (PEBD) [7] – jejímž cílem je efektivně redukovat pool žlučových kyselin a zabránit tvorbě toxických hydrofobních sekundárních žlučových kyselin, může oddálit potřebu TJ, která je v současné době jedinou účinnou léčebnou metodou.
Histologicky je typický obraz obrovskobuněčné hepatitidy. Elektronmikroskopicky je kanalikulární žluč amorfní nebo filamentózní. Spolehlivé určení diagnózy PFIC2 je možné pouze imunohistologickým a/ nebo molekulárně genetickým vyšetřením. Imunohistologicky je BSEP u PFIC2 zpravidla nedetekovatelný. Neléčená PFIC2 obvykle vykazuje rychlejší progresi jaterní fibrózy. Po TJ dochází k ústupu všech příznaků onemocnění. Možnou komplikací PFIC2 je vznik hepatobiliárních malignit [8,9].
V tomto sdělení uvádíme kazuistiky a souborná data šesti dětí s PFIC2 sledovaných na Pediatrické klinice 2. LF UK a FN v Motole, Praha.
Pacienti a metody
Pacienti
Soubor představuje šest dětí europoidního původu narozených v letech 2005 – 2015 nepříbuzným rodičům. Žádný z rodičů nemá klinické ani laboratorní příznaky PFIC2, jen matka prvního pacienta měla od druhého trimestru pruritus bez ikteru, ten po ukončení gravidity vymizel. Sledované děti mají dohromady pět asymptomatických sourozenců.
Imunohistochemické vyšetření
Imunohistochemické vyšetření bylo provedeno ve formolem fixovaných a do parafínu zalitých vzorcích jaterního parenchymu za pomocí automatického barvicího systému Ventana BenchMark ULTRA (Roche, Tucson, Arizona, USA). Histologické řezy tloušťky 4 µm byly inkubovány s primární králičí polyklonální protilátkou anti-ABCB11 (Sigma-Aldrich, St. Louis, Missouri, USA) s následnou vizualizací kitem ultraView Universal DAB Detection Kit (Roche). Jako pozitivní kontrola byly použity řezy zdravé jaterní tkáně.
Mutační analýza
Molekulární vyšetření bylo provedeno na základě písemného souhlasu obou rodičů, kteří podepsali informovaný souhlas před provedením odběru vlastních vzorků krve i krve pacienta. Sekvence genu ABCB11 byla analyzována přímým sekvenováním genomové DNA extrahované z periferních leukocytů. Všech 28 exonů bylo amplifikováno polymerázovou řetězovou reakcí (PCR) s použitím intronických primerů (sekvence jsou dostupné na požádání u autorů). Amplifikované fragmenty byly přečištěny elektroforézou na agarózovém gelu, ze kterého byly extrahovány kitem QIA quick (Qiagen, Hilden, SRN). Sekvenování bylo provedeno pomocí genetického analyzátoru ABI 3130 (Applied Biosystems, Foster City, California, USA). Získané sekvence byly porovnány s referenčními sekvencemi databáze GenBank No. NM_003742 (mRNA) a NT_005403.14 (genomová DNA). Přítomnost zachycených mutací byla nezávisle potvrzena technikou PCR-RFLP. Pozice mutací u dvojitých heterozygotů byly určeny na základě vyšetření rodičů.
Výsledky
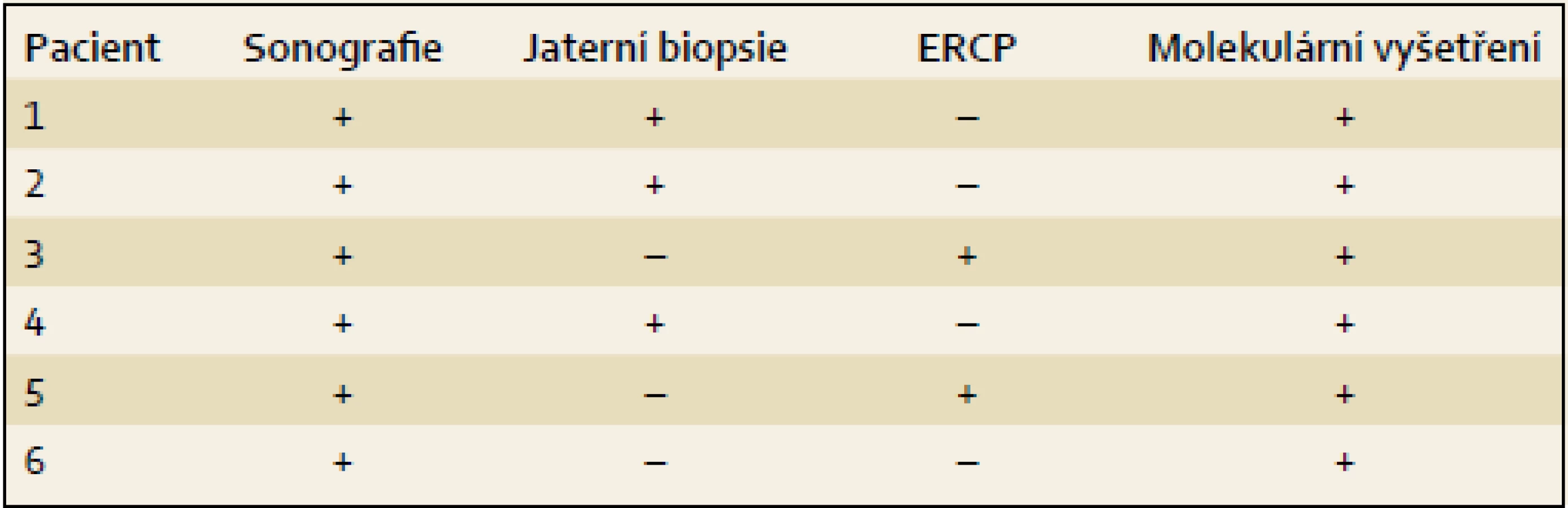
Výsledky jsou uvedeny formou kazuistik. Souborná data jsou prezentována v tab. 1 – 4.

Pacient 1 je chlapec narozený v termínu s porodní hmotností (PH) 3 730 g a porodní délkou (PD) 50 cm, měl výrazný novorozenecký ikterus ovlivněný AB0 inkompatibilitou. V 17. dnu již byla hyperbilirubinemie převážně konjugovaná, provázená normální sérovou aktivitou GGT. V dalších týdnech nález trval (tab. 2), stolice byly mírně hypocholické. Hodnoty α1-fetoproteinu (AFP) byly v normálních mezích. Podrobnější vyšetření stávající NCH vyloučilo její běžné příčiny a sonografie prokázala difuzní jaterní lézi a větší žlučník se sludge (tab. 3). Jaterní biopsie hodnocená na zahraničním pracovišti byla uzavřena s nálezem velkobuněčné transformace hepatocytů a počínající fibrotizace, imunohistochemické vyšetření nezobrazilo BSEP protein. Molekulární vyšetření prokázalo složenou heterozygocii pro dvě mutace v ABCB11 (tab. 4). Bylo zahájeno podávání UDCA a suplementace vitaminů ADEK [10].


Od šestého měsíce byl zřejmý pruritus a neprospívání. Od 12 měsíců stoupaly hladiny AFP. Pravidelná sonografická vyšetření prováděná v odstupu čtyř týdnů prokázala v 17 měsících ložiskovou lézi jater a magnetická rezonance následně zobrazila dvě ložiska (13 a 6 mm) v pravém laloku jaterním. Současně došlo i k prudkému nárůstu AFP na 8 034 µg/ l. PELD skóre (pediatric end stage liver diseases score) bylo 3. V 19 měsících byla provedena TJ (druhý a třetí laterální segment kadaverózního dárce) na zahraničním pracovišti. Ložiska byla vyhodnocena jako hepatocelulární karcinom (HCC). Průběh byl komplikován uzávěrem arteria hepatica, později renálním selháním v souvislosti s chemoterapií. Poté bylo od dalších aplikací upuštěno.
Dítě je imunosuprimováno tacrolimem a mycofenolatem. V dalším průběhu se rozvinul terapií obtížně zvladatelný generalizovaný atopický ekzém. V pěti letech proběhla masivní hemateméza na podkladě hluboké ulcerace v bulbu duodena, která byla po neúspěchu endoskopického řešení ošetřena chirurgicky (dítě pilo výhradně nápoj Kubík).
Nyní je chlapec 10,5letý s funkčním jaterním štěpem, má generalizovaný atopický ekzém provázený pruritem (ŽK jsou mírně zvýšeny na 45 µmol/ l), školní docházka je vedena individuálním přístupem s výbornými výsledky.
Pacient 2 byl chlapec zdravých rodičů. Pro věk matky byla provedena amniocentéza s normálním karyotypem, gravidita ukončena sekcí v 29. týdnu pro preeklampsii – PH 1 180 g, PD 37 cm. Pro časný asfyktický syndrom bylo dítě ventilováno, enterální výživa byla zahájena od devátého dne. Později se na RTG plic rozvinul obraz bronchopulmonální dysplazie.
Dítě mělo novorozenecký ikterus. Hyperbilirubinemie v prvním odběru byla nekonjugovaná, dále již trvale konjugovaná provázená normální aktivitou GGT a vyššími aminotransferázami (tab. 2). Dítě mělo hraniční hepatosplenomegalii, časté a závažně probíhající respirační infekty, neprospívalo, avšak nemělo pruritus a stolice byly hypocholické. V sedmi měsících bylo poprvé přijato na naši kliniku s trvajícím laboratorním nálezem vč. vysoké hodnoty celkových žlučových kyselin (122 µmol/ l), s dobrou proteosyntetickou funkcí jater. AFP byl opakovaně > 200 000 µg/ l. Byly vyloučeny všechny obvyklé příčiny NCH. Sonografie neprokázala ložiskové léze. Jaterní biopsie přinesla nález obrovskobuněčné neonatální hepatitidy s cholestázou. Protein BSEP byl v kanalikulární straně membrány hepatocytu imunohistochemicky prokázán. Molekulární vyšetření však ukázalo přítomnost dvou heterozygotních bodových mutací v pozici trans velmi pravděpodobně podmiňujících PFIC2 (tab. 4). Pacient byl léčen UDCA a suplementací vitaminů ADEK.
Další pneumonie vyústila do respirační insuficience s potřebou umělé plicní ventilace, přidružilo se jaterní selhání a dítě v 11 měsících věku zemřelo. Sekční nález neprokázal ložiska HCC.
Pacient 3 je dívka zdravých rodičů s PH 3 100 g, PD 49 cm. Ikterus na podkladě konjugované hyperbilirubinemie provázený zvýšenými aminotransferázami, normální hodnotou GGT (tab. 2) a hypocholickými stolicemi se projevil až v pěti týdnech. Vyšetřením byly vyloučeny základní příčiny NCH. Byla zahájena terapie UDCA a suplementace vitaminů ADEK. Bilirubin v šesti měsících klesl do normálních hodnot, aminotransferázy byly zvýšené a aktivita GGT byla v normě. Od šesti měsíců měla pruritus a počínající rachitidu, vitamin D byl dále podáván parenterálně. Molekulárně genetické vyšetření prokázalo složenou heterozygocii pro jednu známou patogenní a jednu novou potenciálně patogenní mutaci v ABCB11 (tab. 4). Od tří let stagnovalo růstové tempo, značný pruritus min. reagoval na terapii UDCA a cholestyraminem. Rifampicin byl bez efektu. Od 7,5 let byla přítomna splenomegalie, laboratorní nález se neměnil. V osmi letech se náhle rozvinul těžký ikterus s extrémním pruritem (celkové žlučové kyseliny 428 µmol/ l, z toho UDCA jen 32 %), AFP trvale v normě. Sonografické vyšetření bylo bez ložiskové léze, ale s kolaterálním oběhem. Pruritus nereagoval na sertralin. Prednizon sice vedl k poklesu hladiny bilirubinu, avšak nikoli intenzity pruritu, PEBD nebyla realizována. Nyní (v osmi letech) byla pacientka pro nezvladatelný pruritus, neprospívání a počínající portální hypertenzi (proteosyntetická funkce jater je v normě, je bez průkazu HCC) zařazena na čekací listinu k TJ v IKEM, Praha s PELD skóre 6. Navštěvuje základní školu s výbornými výsledky.
Pacient 4 je dívka narozená v termínu zdravým rodičům s PH 2 750 g a PD 48 cm. Slabý ikterus přítomný již v prvních dnech se zvýraznil v druhém týdnu. Ve čtyřech měsících měla konjugovanou hyperbilirubinemii s hraniční elevací aminotransferáz a normální aktivitou GGT (tab. 2). Stolice byly hypocholické. Jaterní biopsie hodnocená na zahraničním pracovišti ukázala nález obrovskobuněčné transformace hepatocytů, průkaz žlučového pigmentu v hepatocytech a Kupfferových buňkách a počínající fibrotizaci v portálních polích. Imunohistochemická reakce sloužící k průkazu proteinu BSEP v hepatocytech byla negativní. Molekulárně genetické vyšetření prokázalo heterozygotní stav pro dvě patogenní mutace podmiňující PFIC2 (tab. 4). Po zahájení terapie cholestázy klesla hladina bilirubinu na 20 µmol/ l, nevýrazný pruritus byl zřejmý asi od 1,5 roku a tehdy již stagnovalo růstové tempo. Ve dvou letech stoupl celkový bilirubin, dívka měla významnou osteoporózu a po fraktuře tibie dále odmítala chodit. Od tří let byla hladina bilirubinu trvale zvýšena nad 250 µmol/ l a zhoršovala se syntetická funkce jater (PELD 16). Rodiče souhlasili s TJ až ve čtyřech letech dítěte (PELD 24). Dítě psychomotoricky neprospívalo, výška byla výrazně pod třetím percentilem, hodnota AFP byla v normě a nebyla prokázána ložisková léze jater. Za další tři měsíce krvácela pacientka z jícnových varixů. V 4,5 letech byla v IKEM transplantována (druhý a třetí laterální segment kadaverózních jater) a do týdne podstoupila retransplantaci. Imunohistochemické vyšetření v explantovaných játrech opět nezobrazilo BSEP protein – vyšetření již bylo provedeno v IKEM (obr. 1). Nyní, 3,25 roku po transplantaci, je pacientka na terapii tacrolimem a mycofenolatem, prospívá, roste na 10. percentilu, je bez pruritu a její psychomotorický vývoj odpovídá věku.

Pacient 5 je chlapec zdravých rodičů s PH 3 300 g, PD 49 cm. Pacient měl ikterus od prvních dnů života. Ve třech týdnech se již jednalo o konjugovanou hyperbilirubemii se zvýšenými aminotransferázami a normální aktivitou GGT (tab. 2). Chlapec měl výrazně hypocholické stolice, byly vyloučeny obvyklé příčiny NCH. Molekulární vyšetření prokázalo heterozygotní stav pro dvě mutace potvrzující diagnózu PFIC2 (tab. 4). I přes podávání UDCA a vitaminů ADEK se v sedmi měsících přidal pruritus a ve dvou a třech letech vznikly patologické fraktury při těžké osteoporóze, která byla léčena pamidronatem. Již před druhým rokem stagnovala výška, rozvíjelo se chronické jaterní selhání, AFP byl ale trvale v normě a sonografické vyšetření neprokázalo ložiskové léze v jaterním parenchymu. Ve třech letech byl s PELD skóre 12 zařazen na čekací listinu k TJ (druhý a třetí segment jater kadaverózního dárce) realizované za tři měsíce v IKEM. Imunohistochemické vyšetření v explantovaných játrech nezobrazilo BSEP protein. Pacient na imunosupresi tacrolimem výborně psychosomaticky prospívá, akceleruje růst, je bez pruritu. Prodělal CMV infekci a ve čtyřech letech úraz oka.
Pacient 6 je chlapec matky s abúzem pervitinu s heroinem vyléčené z HBV a HCV. Otec je také drogově závislý. Matka byla v graviditě na buprenorpinu, porod byl sekcí v 37. týdnu pro hypoxii plodu, PH 2 300 g, PD 43 cm. Pro novorozenecký abstinenční syndrom byla 12 dnů podávána opiová tinktura, vertikální přenos virových hepatitid nebyl prokázán. Od prvních dnů měl pacient ikterus s klesající intenzitou, v šestém týdnu měl slabou konjugovanou hyperbilirubinemii s normální aktivitou GGT (tab. 2), stolice byly normocholické. V pěti měsících byly aminotransfeázy, GGT i sonografie jater zcela v normě, bilirubin nebyl stanoven. Pruritus byl patrný od osmého měsíce, v jednom roce bylo dítě předáno z ústavní do pěstounské péče. V 16 měsících se při prospívání dítěte objevil ikterus s extrémní gradací pruritu, hepatomegalie bez splenomegalie, stolice byly nadále normocholické. Tehdy byl poprvé vyšetřen na naší klinice. Celkový bilirubin byl 220 µmol/ l, konjugovaný183 µmol/ l, aminotransferázy byly hraničně vyšší, GGT byla v normě, ŽK byly zvýšené na 409 µmol / l a koncentrace AFP byla v mezích normy. Další vyšetření vyloučila obvyklé příčiny cholestázy. Molekulární vyšetření přineslo nález dvou patogenních mutací v ABCB11 (tab. 4). Byla zahájena klasická terapie, po třech měsících je pruritus mírnější, nyní byl přidán rifampicin.
Diskuze
Diferenciální diagnostika cholestázy v novorozeneckém a kojeneckém věku je obtížná pro uniformní klinickou i laboratorní manifestaci jaterních onemocnění. Včasnost stanovení diagnózy a zavedení adekvátní terapie (např. založení portoenteroanastomózy u biliární atrézie, eliminační diety či substituční terapie u metabolických chorob) rozhoduje o prognóze dítěte. Na Pediatrické klinice 2. LF UK a FN v Motole, Praha se od roku 1998 soustřeďují děti s NCH právě za účelem rychlé diferenciální diagnostiky a nastavení správné terapie.
Incidence NCH je 1 : 2 500 živě narozených dětí [11,12]. To v ČR představuje 43 – 44 novorozenců ročně. Etiologicky se jedná o infekční neonatální hepatitidy (v posledních letech je nejčastěji vyvolavatelem cytomegalovirus), z řady metabolických vad je na prvním místě deficit α1-antitrypsinu, endokrinním podkladem jsou hypotyreózy nebo panhypopituitarizmus, z obstrukcí žlučových cest biliární atrézie, cysty choledochu nebo syndrom žlučové zátky. Velkou skupinu NCH tvoří děti s anamnézou sepse, hypoxie, dehydratace. Naopak vzácnou příčinou je Alagilleův syndrom, poruchy metabolizmu ŽK a PFIC.
K úvaze, že příčinou NCH u prezentovaných dětí je právě PFIC2, nás přivedly nízké hodnoty GGT při zvýšených hodnotách konjugovaného bilirubinu, aminotransferáz a vysokých hladinách primárních ŽK, kdy další laboratorní ani sonografické vyšetření provedené k odhalení obvyklých příčin NCH nevedlo k objasnění příčiny choroby. U dvou dětí byla při výrazněji hypocholických stolicích za účelem jednoznačného vyloučení biliární atrézie provedena ERCP s normálním nálezem. Tři děti podstoupily jaterní biopsii doplněnou o imunohistochemický průkaz proteinu BSEP (tab. 3). Všechny děti byly vyšetřeny molekulárně (tab. 4). U posledního z pacientů byla molekulární diagnostika indikována bez předchozích invazivních vyšetření.
Prvním klinickým příznakem PFIC2 byl cholestatický ikterus různé intenzity manifestující se v prvních týdnech života, provázený hypocholickými, event. i normocholickými stolicemi s hraniční hepatomegalií, nebo bez ní. Pruritus se začal manifestovat až kolem šestého měsíce, gradoval a negativně ovlivnil psychomotorický vývoj a kvalitu života dětí. Pravidlem je somatické neprospívání – stagnace růstového tempa, osteoporóza, event. s patologickými frakturami.
K zmírnění jak pruritu, tak i rozvoje fibrotizace jaterního parenchymu je doporučováno podání vysokých dávek UDCA, z čehož má profitovat až třetina pacientů [13]. Eventuálním druhým krokem může být PEBD v situaci, kdy nejsou známky fibrotizace jaterního parenchymu [13]. V našem souboru nebyla nikdy realizována.
Včasná léčba UDCA u našich pacientů rozvoji pruritu nezabránila. Ten je velkým průvodním terapeutickým problémem. Dostupná medikamentózní terapie – kromě UDCA též cholestyramin, rifampicin, sertralin, podávání antihistaminik a lokální terapie – pruritus sice poněkud zmírní, ale nevyřeší. Určitou naději nyní skýtá terapie 4-fenylbutyrátem, který je nyní testován v klinických studiích [14]. Jsou také zprávy o ovlivnění pruritu steroidy [15].
Dle současné natality a předpokládané incidence PFIC by se v ČR ročně měly narodit 1 – 2 děti s PFIC. Počet doposud v ČR diagnostikovaných dětí tomu neodpovídá. Předkládaný soubor šesti dětí představuje pravděpodobně všechny děti diagnostikované v ČR s touto diagnózou (PFIC1 jsme prokázali u jednoho chlapce, PFIC3 u žádného z vyšetřovaných dětí). Lze tedy předpokládat, že jsou diagnostikovány pouze děti s výraznou klinickou manifestací cholestázy vedoucí k jejímu objasnění. Zatímco větší předpokládaný počet jedinců je zřejmě v dětském věku bez klinických obtíží a diagnostice uniká, tak případy, které se manifestovaly již v kojeneckém nebo batolecím věku, mají průběh nepříznivý. Čtyři z šesti dětí podstoupily TJ, u jednoho z nich byl již v 17 měsících prokázán HCC. Jedno dítě zemřelo na jaterní selhání provázené chronickou respirační insuficiencí v 11 měsících. Poslední diagnostikované dítě je sledováno krátce.
Děti s PFIC2 jsou hyperalimentovány, substituovány vitaminy ADEK a jsou jim podávány kalciové preparáty. Přesto je vždy celkový nutriční stav dítěte před TJ alterován. Kvalitní vývoj dítěte lze očekávat až od TJ. Z prezentovaného souboru je pacient 1 již osm let a devět měsíců po transplantaci, trápí jej těžký atopický ekzém a roste pod třetím percentilem. Pacientka 3 byla transplantována před několika dny, pacient 4 je 3,5 roku a pacient 5 je 2,75 roku po transplantaci bez klinických obtíží a došlo k růstovému výšvihu (tab. 1).
Histologický nález PFIC2 v jaterní biopsii provedené v prvních měsících života přibližně odpovídá obrazu idiopatické obrovskobuněčné neonatální hepatitidy (nález balonové degenerace hepatocytů, žlučový pigment v žlučových kanálcích, hepatocytech i Kupfferových buňkách, event. počínající fibrotizace portálních polí). K etiologii léze na podkladě PFIC2 přivede až imunohistochemický průkaz, resp. nezobrazení proteinu BSEP.
Podstatná je diagnostika molekulární, která je neinvazivní a nevyžaduje biopsii. Nález známých patogenních mutací popsaných u PFIC2, případně nález nových zjevně patogenních mutací k diagnóze stačí. Patogenitu dosud nepopsaných missense mutací se doporučuje potvrdit histologicky, typická je téměř kompletní absence BSEP v játrech. Není-li nalezena mutace v jedné či dokonce v obou alelách genu ABCB11 a chybí-li protein BSEP v játrech, je Dg PFIC2 vysoce pravděpodobná i přes (částečně či zcela) negativní nález mutační analýzy. Je-li ale BSEP v kanalikulech exprimován normálně, je Dg PFIC2 nepravděpodobná. Existují ale vzácné výjimky, kdy je nefunkční mutovaný protein exprimovaný [16]. Tuto situaci jsme zaznamenali u našeho pacienta 2. Vedle čtyřnásobného výskytu nejčastější evropské mutace p.Asp482Gly přinesl náš soubor nález sedmi nových mutací. Všichni pacienti jsou složenými heterozygoty pro mutace zděděné od rodičů.
Včasné určení příčiny intrahepatální cholestázy umožňuje adekvátní přístup k danému pacientovi. U PFIC 2. typu lze předpokládat nejen rychlý rozvoj jaterní insuficience v dětském věku, ale i maligní proces v cirhotických játrech. Nejčastěji jsou popisovány HCC [8] již od věku 14 měsíců, popsán byl i hepatoblastom u dvouletého dítěte [17] a cholangiogenní karcinomy u čtyř - a osmiletých dětí [9]. Tumory byly nejčastěji prokazovány až při nekropsii nebo byly náhodným nálezem v explantátu jater. My jsme u jednoho z dětí prokázali dvě ložiska HCC. Náš pacient byl patrně prvním pacientem, u kterého byla transplantace indikována pro maligní komplikaci základní diagnózy. Včasné stanovení diagnózy PFIC2 tedy představuje další aspekt, který je třeba promítnout do postupu při sledování dítěte (pravidelné stanovování koncentrace AFP a provádění sonografických kontrol).
V minulosti byli pacienti s PFIC2 zřejmě chybně vedeni jako idiopatická neonatální hepatitida, o které se tradovalo, že má cca v 15 % případů fatální průběh, neboť základní histologický nález počátečního stadia PFIC2 je s idiopatickou NCH kompatibilní.
Závěr
Včasná správná diagnostika PFIC2 umožní pozitivně ovlivnit průběh onemocnění vč. jeho maligní komplikace. Dostupnost diagnostiky onemocnění i možnost provedení dětské TJ v ČR k tomu dnes dává dostatečné možnosti.
Poděkování
Autoři děkují za technickou asistenci Lucii Budišové.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Doručeno: 20. 11. 2015
Přijato: 27. 11. 2015
MU Dr. Radana Kotalová, CSc.
Pediatrická klinika 2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
radana.kotalova@lfmotol.cuni.cz
Zdroje
1. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012; 36 (Suppl 1): S26 – S35. doi: 10.1016/ S2210-7401(12)70018-9.
2. Strautnieks SS, Bull LN, Knisely AS et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998; 20(3): 233 – 238.
3. Jansen PL, Müller M. The molecular genetics of familial intrahepatic cholestasis. Gut 2000; 47(1): 1 – 5.
4. Gerloff T, Stieger B, Hagenbuch B et al. The sister of P-glycoprotein represents thecanalicular bile salt export pump of mammalian liver. J Biol Chem 1998; 273(16): 10046 – 10050.
5. Davis RA, Miyake JH, Hui TY et al. Regulation of cholesterol-7alpha-hydroxylase: BAREly missing SHP. J Lipid Res 2002; 43(4): 533 – 543.
6. Thompson R, Strautnieks SS. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis 2001; 21(4): 545 – 550.
7. Kaliciński PJ, Ismail H, Jankowska I et al. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg 2003; 13(5): 307 – 311.
8. Knisely AS, Strautnieks SS, Meier Y et al. Hepatocellular carcinoma in ten children under five years old with bile salt export pump deficiency. Hepatology 2006; 44(2): 478 – 486.
9. Scheimann AO, Strautnieks SS, Knisely ASet al. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr 2007; 150(5): 556 – 559.
10. Kotalová R, Cebecauerová D, Knisely AS et al. Progresivní familiární intrahepatální cholestáza – manifestace a diagnostika v kojeneckém věku. Čes-slov Pediat 2006; 61(4): 200 – 206.
11. Balistreri WF. Neonatal cholestatis. J Pediatr 1985; 106(2): 171 – 184.
12. McKiernan PJ. Neonatal cholestasis. Semin Neonatol 2002; 7(2): 153 – 165.
13. Davit-Spraul A, Gonzales E, Baussan C et al. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis 2009; 4 : 1. doi: 10.1186/ 1750-1172-4-1.
14. Naoi S, Hayashi H, Inoue T et al. Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. J Pediatr 2014; 164(5): 1219 – 1227. doi: 10.1016/ j.jpeds.2013.12.032.
15. Engelmann G, Wenning D, Herebian D et al. Two case reports of successful treatment of cholestasis with steroids in patients with PFIC-2. Pediatrics 2015; 135(5): e1326 – e1332. doi: 10.1542/ peds.2014 – 2376.
16. Strautnieks SS, Byrne JA, Pawlikowska Let al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008; 134(4): 1203 – 1214. doi: 10.1053/ j.gastro.2008.01.038.
17. Richter A, Grabhorn E, Schulz A et al. Hepatoblastoma in a child with progressive familial intrahepatic cholestasis. Pediatr Transplant 2005; 9(6): 805 – 808.
Štítky
Detská gastroenterológia Gastroenterológia a hepatológia Chirurgia všeobecnáČlánok vyšiel v časopise
Gastroenterologie a hepatologie

2015 Číslo 6
- Těhotenství a idiopatické střevní záněty – terapie zlepšuje stav a je bezpečná
- Vliv těhotenství na klinickou aktivitu Crohnovy nemoci
- Medikace u IBD v těhotenství
- Adherence a kvalita života adolescentních pacientů s IBD
- Adherence pacientů se střevním zánětlivým onemocněním
Najčítanejšie v tomto čísle
- Ursodeoxycholová kyselina (Ursosan® tobolky)
- Výlučná enterální výživa – léčba první volby Crohnovy choroby u dětí
- Výsledky transplantácií pečene u slovenských detí
- Význam genetického vyšetrenia u detí s idiopatickou chronickou pankreatitídou