
Nové poznatky v patogenezi Crohnovy choroby
New knowledge of the pathogenesis of Crohn’s disease
Crohn’s disease is a complex chronic inflammatory disease of the gastrointestinal tract with multifactorial pathogenesis. Over the recent years, there has been rather a sharp increase in the incidence of Crohn’s disease and, even though this disease had been known for some time, the cause remains unknown. Studies exploring genetic basis of Crohn’s disease have provided new knowledge of the pathogenesis of this disease, suggesting that this may be associated with a failure of mechanisms behind symbiosis of gut microflora and intestinal mucosal immune system. Crohn’s disease seems to be caused by inadequate immune response to intestinal flora in genetically predisposed individuals. Crohn’s disease has been linked to a number of genes. Many of them are related to the modulation of non-specific immune response, defects of which are considered to be key in Crohn’s disease pathogenesis. The aim of this review paper is to summarize the new knowledge on the pathogenesis of Crohn’s disease at the level of polymorphisms of the NOD2, ATG16L1 genes and the IL23-Th17-lymfocytes signalling pathway genes and to consider further research directions in this disease.
Key words:
Crohn’s disease – pathogenesis – NOD2 – ATG16L1 – IL23-Th17-lymphocytes
Autoři:
B. Ambrůzová 1; M. Rédováihash2 1,2,3 1,2,3
Působiště autorů:
Advanced Cell Immunotherapy Unit, Lékařská fakulta MU Brno, vedoucí doc. MUDr. Dalibor Valík, Ph. D.
1; Klinika komplexní onkologické péče, MOÚ Brno, přednosta prof. MUDr. Rostislav Vyzula, CSc.
2; Výzkumná skupina Molekulární onkologie II – solidní nádory Středoevropského technologického institutu Brno, vedoucí RNDr. Ondřej Slabý, Ph. D.
3; Gastroenterologické oddělení, MOÚ Brno, vedoucí MUDr. Milana Šachlová, CSc. et Ph. D.
4
Vyšlo v časopise:
Vnitř Lék 2012; 58(4): 291-298
Kategorie:
Přehledné referáty
Souhrn
Crohnova nemoc je komplexní chronické zánětlivé onemocnění trávicí soustavy, na jehož patogenezi se podílí celá řada faktorů. Incidence Crohnovy nemoci v posledních letech zaznamenala poměrně prudký nárůst, a přestože je tato nemoc známa již delší dobu, její příčinu se doposud nepodařilo odhalit. Nové poznatky v patogenezi onemocnění přinesly studie zabývající se genetickou podstatou Crohnovy nemoci. Výsledky těchto studií poukazují na selhání mechanizmů symbiózy střevní mikroflóry a střevního slizničního imunitního systému. Zdá se, že Crohnova nemoc vzniká nepřiměřenou reakcí imunitního systému na střevní mikroflóru u geneticky predisponovaných jedinců. S Crohnovou nemocí byla asociována celá řada genů. Mnohé z nich souvisejí s funkcí nespecifické imunitní odpovědi, jejíž nedostatky jsou v dnešní době považovány za klíčové v patogenezi Crohnovy nemoci. Cílem tohoto přehledového článku je shrnout nové poznatky v patogenezi Crohnovy nemoci na úrovni polymorfizmů v genech NOD2, ATG16L1 a v genech signální dráhy IL23-Th17-lymfocyty a zmínit další směry výzkumu v problematice tohoto onemocnění.
Klíčová slova:
Crohnova nemoc – patogeneze – NOD2 – ATG16L1 – IL23-Th17-lymfocyty
Úvod
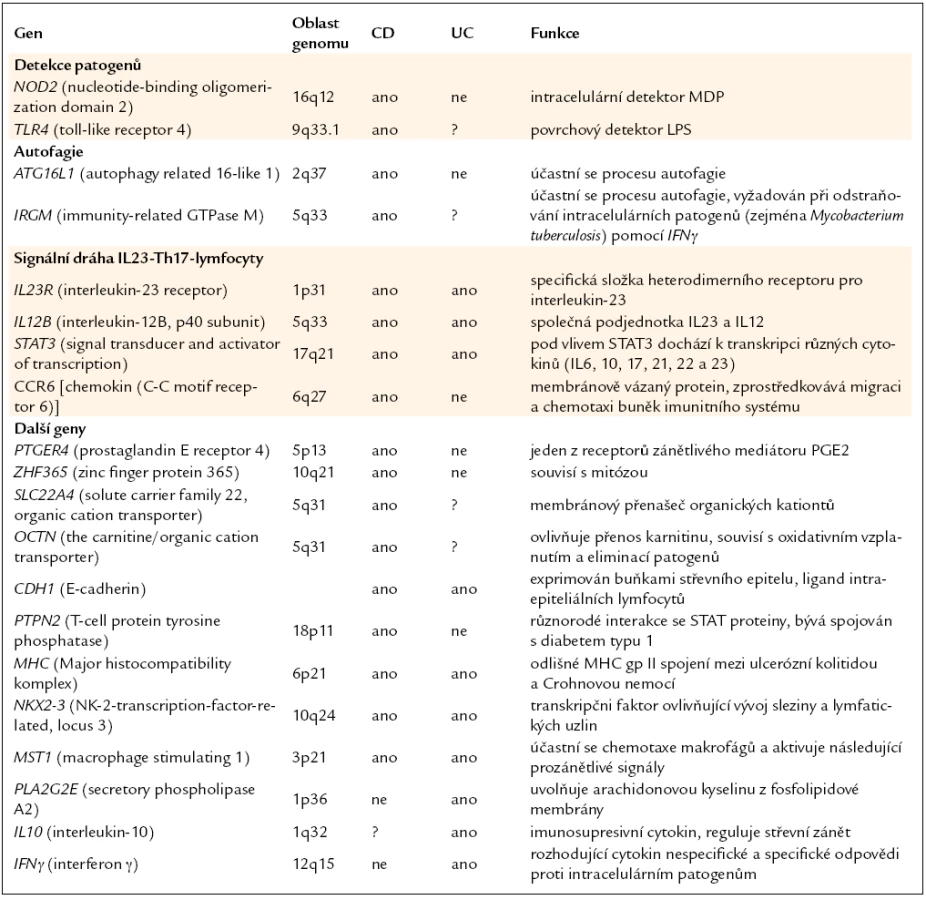
Crohnova nemoc patří mezi nespecifické střevní záněty. Jedná se o chronické zánětlivé onemocnění trávicí soustavy. Přestože je tato choroba známa již několik desítek let, etiologii onemocnění se stále nepodařilo objasnit a ani patogenetické mechanizmy nejsou uspokojivě popsány. Dříve se předpokládalo, že je Crohnova nemoc způsobena agresivní zánětlivou odpovědí a nekontrolovanou produkcí prozánětlivých mediátorů, což bylo později částečně potvrzeno úspěšným použitím biologické terapie namířené proti TNFα (tumor necrosis factor α) [1]. Hromadící se důkazy z posledních let však naznačují, že nemoc vzniká v důsledku nepřiměřené reakce na střevní mikroflóru u geneticky predisponovaných jedinců [2]. Hlubší porozumění patogenezi tohoto onemocnění přinesly v poslední době tzv. GWAS (genome-wide association studies), na jejichž základě byla identifikována řada genů asociovaných s touto nemocí [3–6].V současné době je potvrzeno více než 30 genů majících spojitost s patogenezí Crohnovy nemoci a je pravděpodobné, že se jejich počet bude zvyšovat [7]. Některé z nich jsou shrnuty v tab. 1. Crohnova nemoc je tedy komplexní onemocnění s polygenním základem. Je však zřejmé, že pro vznik a vývoj nemoci je nutná nejen přítomnost genetických predispozic, ale jejich kombinace s vlivy vnějšího prostředí (obr. 1). Bylo navrženo několik teorií, jejichž cílem je odhalit takový faktor, popřípadě kombinaci více faktorů, jejichž působení by mělo vliv na vznik a průběh Crohnovy nemoci. Nejčastěji bývá v této souvislosti zmiňována hygienická hypotéza, která předpokládá, že na vzniku nemoci má značný podíl nedostatečný kontakt s enterickými patogeny v dětství. Takový jedinec je pak v dospělosti přecitlivělý na setkání s běžnou infekcí a může se u něj Crohnova nemoc vyvinout. Svou roli podle hypotézy sehrávají přehnané hygienické návyky. Teorie také zohledňuje rodinné zázemí, způsob porodu, počet sourozenců, zda byla prodělána infekce Helicobacter pylori, kontakt s domácími zvířaty a další. Z výsledků epidemiologických studií také vyplývá, že Crohnovou nemocí jsou častěji postiženy osoby žijící ve vyspělých hustě osídlených oblastech, na rozdíl od venkovské populace, kde je Crohnova nemoc méně častá [8].

Četné důkazy upozorňují na pozměněnou odpověď imunitního systému na mikrobiální motivy jako na jeden z nejdůležitějších příspěvků v patogenezi onemocnění [9]. To bylo prokázáno dalšími studiemi, které ukazují, že chronický zánět typický pro Crohnovu nemoc se odvíjí od přítomnosti střevní mikroflóry [10,11]. Bakteriální druh, který by mohl být příčinou vzniku Crohnovy nemoci, však doposud nebyl identifikován. Podařilo se zjistit, že tenké střevo, obzvláště ileum, pacientů s Crohnovou nemocí je kolonizováno patogenním druhem bakterie Escherichia coli. Bakterie tohoto druhu jsou schopny adherovat ke střevním epiteliálním buňkám a pronikat do nich. Nedávná Chassainova studie se zabývá způsobem, jakým tyto a jiné invazivní bakterie mohou způsobovat vznik časných lézí v ileu pacientů s Crohnovou nemocí [12].
Střevní mikroflóra a také potravinové antigeny jsou v neustálém kontaktu se střevní sliznicí. Jednovrstevný epitel, který odděluje dutinu střeva a střevní slizniční imunitní systém, funguje jako fyzikální bariéra proti potravinovým antigenům a mikrobům, ale aktivně se také podílí na zpracování případných antigenů a následné regulaci imunitní reakce [13]. Enterocyty plní funkci vstřebávání vody a živin, ale např. pohárkové buňky produkují hlen, který vytváří vrstvu na povrchu epitelu, a omezují tak nadměrný kontakt sliznice se střevní mikroflórou [14]. Některé buňky střevního epitelu produkují defenziny. Defenziny fungují jako endogenní antibiotika s mikrobicidní aktivitou proti grampozitivním a gramnegativním bakteriím. Přirozená produkce defenzinů a dalších antimikrobiálních peptidů v trávicím traktu pomáhá omezovat invazi a adherenci komenzálních a patogenních bakterií a napomáhá regulovat složení střevní mikroflóry. V tenkém střevě je, na rozdíl od tlustého střeva, mnohem méně bakterií. Podílí se na tom zřejmě nadprodukce α-defenzinů Panethovými buňkami [15]. Defenziny jsou v Panethových buňkách uloženy v sekrečních váčcích, k jejichž uvolnění do lumen střeva dochází až po stimulaci bakteriálními produkty. Kromě α-defenzinů existují i β-defenziny uvolňované specializovanými epiteliálními buňkami tlustého střeva. Za normálních okolností je konstitutivně produkován β-defenzin HBD1. Jen během zánětu nebo infekce dochází k syntéze i jiných defenzinů, např. inducibilního β-defenzinu HBD2 [16].
Dalším specializovaným typem enterocytů jsou M buňky. Ty pohlcují antigeny z lumen střeva a přenášejí je na opačnou stranu, kde případné antigeny předávají ke zpracování dendritickým buňkám, nebo jsou přímo rozpoznány antigenně specifickými receptory B-lymfocytů [17]. M buňky se vyskytují v těsné blízkosti Peyerových plátů. V Peyerových plátech jsou T - i B-lymfocyty, dendritické buňky a makrofágy. Ve sliznici se nacházejí dendritické buňky, které vysílají své výběžky mezi epiteliální buňky až do lumen zažívacího traktu a aktivně vychytávají antigeny. Přímo v epiteliální vrstvě střeva jsou přítomny i tzv. intraepiteliální T-lymfocyty morfologicky podobné dendritickým buňkám, které monitorují poškozené buňky střevního epitelu [18]. Slizniční lymfocyty ovlivňují funkci epiteliálních buněk a epiteliální buňky mohou expresí MHC gp II fungovat jako buňky prezentující antigen a ovlivňovat imunokompetentní buňky [19]. Je zřejmé, že jakákoli změna v rozpoznávání bakterií a nedostatky v procesech vedoucích k jejich zneškodnění mohou způsobit selhání přirozené symbiózy mezi slizničním imunitním systémem a střevní mikroflórou [20,21].
Detekce patogenů, NOD2
V roce 2001 byl jako první gen vykazující predispozici ke vzniku Crohnovy choroby identifikován NOD2 (nucleotide-binding oligomerization domain containing 2, známý též jako IBD1 nebo CARD15) [22,23]. NOD2 se uplatňuje jako vnitrobuněčný receptor MDP (muramyl dipeptid) [24]. MDP je odvozen od peptidoglykanu, který tvoří strukturu buněčné stěny grampozitivních i gramnegativních bakterií. NOD2 je exprimován monocyty, makrofágy, dendritickými buňkami, buňkami střevního epitelu (obzvláště Panethovými buňkami), ale i Treg (T regulační) lymfocyty. NOD2 za normálních okolností odpovídá na setkání s MDP aktivací NF-κB (nuclear factor of κ light polypeptide gene enhancer in B-cells) a MAPK (mitogen activated protein kinase), což vede k produkci prozánětlivých cytokinů a antimikrobiálních peptidů [25]. Charakteristickým znakem NOD2 jsou repetice bohaté na leucin v C koncové oblasti proteinu, které se účastní vnitrobuněčného rozpoznávání patogenů [26]. Změny v genu NOD2 vyskytující se v této oblasti jsou reprezentovány 3 polymorfizmy (R702W, G908R, 1007fs) [27]. Přestože je NOD2 v souvislosti s Crohnovou chorobou nejvíce prostudovaný, stále není úplně jasné, jakým způsobem polymorfizmy v tomto genu ovlivňují náchylnost ke vzniku Crohnovy nemoci. U nositelů polymorfizmu v genu NOD2 byla zjištěna snížená aktivace NF-κB po stimulaci NOD2 bakteriálním peptidoglykanem [28] a také snížená produkce α-defenzinů Panethovými buňkami [29]. Pokles α-defenzinů, např. HD5 a HD6, byl prokázán u pacientů s ileální formou Crohnovy nemocí, kteří nesli polymorfizmus v genu NOD2, zatímco hladiny TNFα a IL8 byly nezávislé na přítomnosti polymorfizmu. Exprese α-defenzinů v případě pacientů s Crohnovou nemocí tlustého střeva zůstala nezměněna. Funkčním následkem nízké hladiny α-defenzinů byla zeslabená antibakteriální obrana. Přestože přibližně jen 1/3 všech pacientů nese polymorfizmus v genu NOD2, snížená produkce α-defenzinů byla nalezena v podstatě u všech pacientů. Musí tedy existovat ještě další mechanizmus. Diferenciace Panethových buněk je řízena Wnt signalizační dráhou, která je trans-dukována prostřednictvím β-cateninu a transkripčního faktoru Tcf-4. Tato dráha udržuje progenitorové buňky v nediferencovaném stavu, ale zároveň indukuje maturaci Panethových buněk v intestinálních kryptách. Narušení této dráhy bylo pozorováno u pacientů s ileální formou Crohnovy nemoci. Hladiny Tcf-4 byly sníženy stejně jako hladiny defenzinů HD5 a HD6. Podobné snížení ale nebylo zřejmé u pacientů s postižením tlustého střeva. Bylo také zjištěno, že spojení mezi poklesem defenzinů Panethových buněk a Tcf-4 je nezávislé na genotypu NOD2 [30]. Pozorován byl také pokles β-defenzinů. Hladiny HBD1 i HBD2 jsou nižší v tkáni tlustého střeva postiženého Crohnovou nemocí. Tento pokles je ale zřejmě způsoben sníženým počtem genů pro β-defenziny a nesouvisí s přítomností polymorfizmu v genu NOD2. Vadná produkce defenzinů vede k postupnému přemnožení bakterií, jejich pomalé invazi střevní sliznicí a následně k imunologické odpovědi na střevní mikroflóru. Určité probiotické kmeny mají schopnost stimulovat produkci defenzinů. Užití probiotik má pozitivní vliv na průběh ulcerózní kolitidy, ale v případě Crohnovy nemoci mají probiotika omezené užití a jsou spíše méně užitečná [31].
Nedávno bylo také dokázáno, že N-glykolovaný MDP, který se vyskytuje u mykobakterií a některých aktinomycet, mnohem účinněji aktivuje NF-κB než běžně se vyskytující N-acetylovaný MDP. Je tak možné, že polymorfizmy v genu NOD2 znemožňují správnou identifikaci mykobakterií. Pokud mykobakterie nejsou imunitním systémem rozpoznány, lidské tělo nedokáže infekci účinně čelit. Přestože je souvislost mezi Crohnovou nemocí a mykobakteriální infekcí známa už delší dobu [32], není stále jasné, jestli je přítomnost mykobakterií příčinou, nebo důsledkem tohoto onemocnění [33].
Rahmanova studie se zabývá vztahem NOD2 a Treg lymfocytů. Bylo dokázáno, že tyto lymfocyty jsou po stimulaci MDP chráněny před apoptózou zprostředkovanou ligandy Fas receptoru. Tato ochrana ale nebyla patrná u MDP-stimulovaných Treg buněk pacientů s Crohnovou chorobou, u kterých byl prokázán polymorfizmus v genu NOD2. Studie tak odhalila, že jednou z funkcí, kterou může NOD2 v Treg lymfocytech zastávat, je jejich ochrana proti apoptóze, k níž může docházet v prostředí bohatém na ligandy Fas receptoru (např. TNFα), jako je tomu např. v místě zaníceného střevního epitelu. To by mohlo být důvodem sníženého množství Treg lymfocytů, které bylo již dřív pozorováno u pacientů s Crohnovou nemocí [34].
Studie jednovaječných dvojčat prokázala, že v 63,6 % trpí obě dvojčata Crohnovou nemocí [35], což ukazuje na významný podíl genetických predispozic pro vznik Crohnovy nemoci. Polymorfizmy v genu NOD2 se vyskytují s vyšší frekvencí u evropské populace, kde nese nejméně jeden z výše jmenovaných 3 polymorfizmů přibližně 30 % všech pacientů [36]. Četnost jednotlivých variant se ale značně liší napříč populacemi. V České republice se u 46 % pacientů vyskytuje alespoň jedna forma genu NOD2 a nejsilněji je s Crohnovou nemocí asociován polymorfizmus 1007fs. 42 % pacientů, kterým byla Crohnova nemoc diagnostikována v dětství, nese právě tuto variantu genu NOD2. Frekvence 1007fs v Čechách je 20 %, zatímco v severní Evropě jsou tyto hodnoty nižší (2,7–3 % Norsko, 4,8 % Finsko) [37]. Jak již bylo zmíněno, přítomnost polymorfizmu v genu NOD2 vykazuje spojitost s postižením ilea a s tvorbou fibrostenóz [38].
Detekce patogenů, TLR
TLR (toll-like receptors) fungují jako vnější receptory některých bakteriálních znaků (obr. 2) a jsou exprimovány na povrchu monocytů, makrofágů, dendritických a epiteliálních buněk. Dendritické buňky a makrofágy aktivované TLR produkují prozánětlivé cytokiny a chemokiny a exprimují vysoké hladiny kostimulačních molekul. Na rozdíl od toho signalizace prostřednictvím TLR ve střevních epiteliálních buňkách zabraňuje rozvoji zánětlivé odpovědi a podporuje navození homeostázy [39]. Střevní epiteliální buňky za normálních okolností exprimují ve větší míře TLR3 a TLR5, zatímco TLR2 a TLR4 jsou na povrchu těchto buněk přítomny v mnohem menších množstvích [40]. U pacientů s nespecifickými střevními záněty vykazují buňky střevního epitelu změny v expresi TLR3 a TLR4. V aktivní fázi onemocnění je patrné menší množství TLR3 pouze v případě Crohnovy nemoci. Na rozdíl od toho je zvýšená exprese TLR4 jak v případě Crohnovy nemoci, tak ulcerózní kolitidy. Exprese TLR2 a TLR5 zůstává nezměněná [41]. TLR jsou exprimovány i na povrchu Th-lymfocytů, jejich funkce ale není detailně prozkoumána. Přitom TLR a TCR (T cell receptor) sdílejí podobnou signalizační dráhu, např. MAPK. Výsledky Gonzales-Navajasovy studie prokázaly, že TLR4 exprimované na povrchu efektorových Th-lymfocytů upravují po stimulaci LPS (lipopolysacharid) následnou aktivaci TCR a omezují zánětlivé procesy [42].

V souvislosti s Crohnovou nemocí byl potvrzen polymorfizmus v genu pro TLR4. Tento polymorfizmus je charakteristický záměnou asparaginu na 299. pozici za glycin (D299G) [43]. TLR4 váže kromě jiných ligandů hlavně LPS, který je hlavní složkou vnější membrány gramnegativních bakterií. Arbour et al prokázali, že nositelé polymorfizmu Asp299Gly mají oslabenou imunitní odpověď na inhalovaný LPS [44]. V jiné studii, kde byly LPS stimulované vzorky plné krve, byla prokázána vyšší prozánětlivá odpověď za uvolnění velkého množství TNFα [45]. Důležité také je, že mezi signálními drahami NOD2 a TLR existuje propojení. Zdá se, že za normálních okolností NOD2 negativně reguluje aktivitu TLR2. V případě chybějícího NOD2 nebo v přítomnosti polymorfizmu souvisejícího s Crohnovou nemocí (1007fs) v genu NOD2 může docházet ke ztrátě této regulace, tím pádem k intenzivnější aktivaci NF-κB prostřednictvím TLR2 a k nadměrné zánětlivé odpovědi Th1-lymfocytů [46]. Později bylo dokázáno, že takto NOD2 reguluje aktivitu i TLR4 [47].
Autofagie, ATG16L1
Autofagie je proces, díky kterému má buňka schopnost trávit a recyklovat své vlastní organely v období hladovění. Pro Crohnovu nemoc je však důležitější, že autofagie hraje významnou roli v boji proti vnitrobuněčným mikroorganizmům, které pronikly do cytosolu. Autofagie je důležitou součástí nespecifické imunitní odpovědi z toho důvodu, že odstraňuje intracelulární bakterie a viry, ale je to také proces spojený se specifickou imunitou, protože degradované vnitrobuněčné proteiny může buňka vystavit na svém povrchu jako antigeny pomocí MHC gp II. Ty pak mohou být rozpoznány Th-lymfocyty a dendritickými buňkami. Tento způsob prezentace antigenů propojující autofagii a MHC gp II je nejlépe popsán v souvislosti s profesionálními antigen-prezentujícími buňkami, jako jsou např. dendritické buňky nebo makrofágy. Přesto může mít význam i v epiteliálních buňkách, které exprimují MHC gp II v místě zánětu, a stávají se tak neprofesionálními antigen-prezentujícími buňkami [48].
Autofagie je regulována různými podněty, např. cytokinovými receptory nebo TLR, a její průběh je znázorněn na obr. 3. K navození autofagie dochází také prostřednictvím NOD2 v přítomnosti MDP. Bylo však zjištěno, že varianty genu NOD2 spojované s Crohnovou nemocí narušují průběh autofagie [49].

Několik GWAS potvrdilo spojitost mezi genem ATG16L1 a Crohnovou nemocí. ATG16L1 je jedním z klíčových proteinů potřebných pro správné vytvoření autofagozomu. Výsledky analýz naznačují, že riziko vzniku Crohnovy nemoci souvisí s polymorfizmem T300A [27]. Pouze tato varianta ATG16L1 má za následek specifickou poruchu autofagie v přítomnosti bakterií [50] a zeslabuje antibakteriální funkce v epiteliálních buňkách odvíjející se od NOD2 [49]. U myší s defektními ATG16L1 proteiny byly pozorovány změny v sekreci IL1β [51]. Cadwell et al ve své studii zaznamenali abnormality v Panethových buňkách, jako jsou např. degenerující mitochondrie a ztráta lysozymových granulí [52].
V posledních letech bylo navození autofagie často považováno za následek aktivace nespecifické imunitní odpovědi. Existují však přesvědčivé důkazy, že mezi autofagií a nespecifickou imunitou je vzájemný vztah. Cytokiny specifické i nespecifické imunity různými způsoby ovlivňují autofagii. Předpokládá se také, že autofagie reguluje sekreci cytokinů u pacientů s Crohnovou nemocí [53].
Signální dráha IL23-Th17-lymfocytů
Tradičně bývá Crohnova nemoc spojována s převažujícím podtypem Th1-lymfocytů, a tím pádem zvýšeným množstvím cytokinů, které tyto lymfocyty produkují. Hlavním z nich je IFNγ (interferon γ), jehož úkolem je stimulace makrofágů k fagocytóze, a tedy obrana proti intracelulárním mikrobům [54]. Tento koncept byl změněn poté, co byly popsány Treg lymfocyty a prozánětlivé Th17-lymfocyty. Již několik studií prokázalo, že na patogenezi Crohnovy nemoci se kromě Th1 buněk podílejí i Th17-lymfocyty [55]. Populace Th17-lymfocytů je charakteristická produkcí prozánětlivých cytokinů IL17A, IL17F, IL21, IL22, IL26, TNFα, IFNγ a CCL20 [56]. Zvýšené hladiny IL17A, IL17F, IL22 byly potvrzeny u pacientů s aktivní formou Crohnovy choroby [57].
Th1-lymfocyty diferencují pod vlivem IL12. Tento prozánětlivý cytokin byl dlouho považován za rozhodující v patogenezi Crohnovy nemoci. Avšak zjištění, že IL23, který je třeba pro diferenciaci Th17-lymfocytů, je heterodimerní cytokin skládající se ze specifické p19 podjednotky a p40 řetězce, který také tvoří součást IL12 [58], nutně vede k přehodnocení jejich vzájemných vztahů v patogenezi Crohnovy nemoci. Dalším důkazem je skutečnost, že užití monoklonálních protilátek (ustekinumab) proti p40 podjednotce, kterou sdílí IL12 a IL23, vyvolalo klinickou odpověď u pacientů s Crohnovou nemocí [59]. IL23 je secernován dendritickými buňkami a makrofágy po setkání s antigenem a přispívá k proliferaci Th17-lymfocytů. Signalizace zprostředkovaná IL23 je zajištěna jeho interakcí s receptorem exprimovaným na povrchu Th17-lymfocytů, který je také heterodimerní a skládá se ze 2 podjednotek – IL23R a IL12RB1 [2].
Na základě GWAS byl v souvislosti s Crohnovou nemocí objeven polymorfizmus v genu kódujícím IL23R (interleukin 23 receptor) [55]. Bylo také prokázáno, že kromě IL23R vykazují souvislost s patogenezí Crohnovy nemoci polymorfizmy i v dalších genech zapojených v diferenciaci Th17-lymfocytů. Jsou to IL12B, JAK2 a STAT3, u kterých bylo později prokázáno i spojení s ulcerózní kolitidou [27].
Aktivita Th17-lymfocytů má vliv na biologickou funkci střevní bariéry (obr. 4), protože buňky střevního epitelu exprimují receptory pro některé interleukiny uvolňované Th17 buňkami. IL17A, IL21 a IL22 podporují hromadění prozánětlivých buněk ve střevní sliznici díky jejich schopnosti zvyšovat syntézu chemoatraktantů epiteliálními buňkami a adhezivních molekul (např. ICAM-1) endoteliálními buňkami [60]. IL21 a IL22 stimulují fibroblasty k tvorbě tkáňových metaloproteináz, tedy enzymů, které mohou přispívat k poškození a remodelaci tkáně [61,62]. IL17A a IL22 dále podporují syntézu antibakteriálních proteinů (např. defenzinů) epiteliálními buňkami [63], a tak mají kromě svých prozánětlivých vlastností i pozitivní vliv na funkci střevní bariéry. IL17 udržuje integritu střevní bariéry modulací „tight junctions“ (těsných spojení) mezi enterocyty a IL22 zvyšuje migrační a proliferační potenciál intestinálních epiteliálních buněk [57]. Dalším cytokinem produkovaným Th17-lymfocyty je IL26. Na rozdíl od proliferativních účinků IL22, IL26 snižuje buněčnou proliferaci [64], a tudíž potenciálně zhoršuje hojení střevních poranění. Th17-lymfocyty dále uvolňují chemokin CCL20. Receptorem tohoto chemokinu je CCR6, který je exprimován na povrchu Th17-lymfocytů. CCL20 tak může přispívat k udržování zánětu, protože autokrinně reguluje diferenciaci Th17-lymfocytů [57].
![Diferenciace a funkce Th17-lymfocytů (upraveno podle [60]). Th17-lymfocyty diferencují pod vlivem TGFβ v kombinaci s IL1β, IL6 a na rozdíl od Th1-lymfocytů nevyžadují pro diferenciaci své efektorové cytokiny, pro jejich diferenciaci není nutná přítomnost IL17. IL21 reguluje diferenciaci Th17 buněk autokrinním způsobem. IL23 slouží k amplifikaci zánětlivé odpovědi odvozené od Th17-lymfocytů. Th17-lymfocyty produkují prozánětlivé cytokiny, ale i látky ovlivňující funkci střevní bariéry.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/17b428433bf5b8cce48fbdbfdad0f5aa.png)
T regulační lymfocyty
Podstatnou roli v patogenezi Crohnovy nemoci sehrávají také supresivní Treg lymfocyty. Jak již bylo výše zmíněno, při diferenciaci Th17-lymfocytů hraje roli TGFβ. Tento cytokin je ale také nezbytný pro diferenciaci Treg. Nízká koncentrace TGFβ navozuje diferenciaci do Th17 buněčné linie, zatímco vysoké hladiny tohoto cytokinu podporují vznik Treg [65]. Zajímavé je i to, že v přítomnosti prozánětlivého IL6 může docházet k přeměně Treg na Th17-lymfocyty [66]. Vzájemný vztah mezi Treg a Th17 podporuje také fakt, že IL2, který je růstovým faktorem pro Treg, inhibuje vývoj Th17-lymfocytů [67].
Shrnutí
Je zřejmé, že v patogenezi Crohnovy nemoci se uplatňují změny na úrovni imunitního systému. Důležitým mechanizmem je s největší pravděpodobností selhání koexistence střevní mikroflóry a střevního slizničního imunitního systému. Složení střevní mikroflóry není stálé a mění se pod vlivem vnějších podmínek. Podle nových poznatků vzniká zánět typický pro Crohnovu nemoc nepřiměřenou reakcí na střevní mikroflóru u geneticky predisponovaných jedinců. Nejdéle známým genem vykazujícím predispozici ke vzniku Crohnovy nemoci je NOD2. Je exprimován v různých typech buněk a zastává v organizmu komplexní úlohu – ovlivňuje sekreci cytokinů, průběh autofagie a apoptózy. Je tedy těžké jednoznačně posoudit jeho význam v patogenezi onemocnění. U nositelů polymorfizmů v genu NOD2 byla zjištěna snížená produkce α-defenzinů Panethovými buňkami. Tento typ buněk je také postižen v souvislosti s polymorfizmy v genu ATG16L1, který se podílí na autofagii. S Crohnovou nemocí byl také asociován polymorfizmus v genu pro TLR4. TLR4 podobně jako NOD2 funguje jako detektor bakteriálních znaků. Studie zabývající se polymorfizmem v TLR4 ale podávají rozporuplné výsledky. Dále bylo zjištěno, že kromě Th1-lymfocytů, které byly dlouhou dobu považovány za klíčové v patogenezi Crohnovy nemoci, se v průběhu nemoci uplatňuje i poměrně nedávno objevená populace Th17-lymfocytů. Th17-lymfocyty produkují prozánětlivé mediátory (TNFα, IFNγ), ale i cytokiny, které ovlivňují funkci střevní bariéry. Navíc kromě změn v imunitním systému byly pozorovány i poruchy střevního epitelu, např. narušení vrstvy hlenu, dysregulace těsných spojení mezi buňkami epitelu, zvýšená propustnost epitelu či zvýšená bakteriální přilnavost k epiteliálním buňkám. Není však jasné, zda jsou tyto změny primární příčinou vzniku zánětu, nebo jestli vznikají až v reakci na již probíhající zánět. Velkou výzvou do budoucnosti zůstává zjistit, jaké další funkční následky mají změny v genech souvisejících s Crohnovou nemocí a do jaké míry přispívají k patogenezi onemocnění.
Práce byla podpořena grantem Ministerstva zdravotnictví ČR IGA IGA NS/10352-3/2009 a výzkumným záměrem MZ0MOU2005.
RNDr. Ondřej Slabý, Ph.D.
www.mou.cz
e-mail: slaby@mou.cz
Doručeno do redakce: 12. 7. 2011
Přijato po recenzi: 5. 1. 2012
Zdroje
1. Kozuch PL, Hanauer SB. Treatment of inflammatory bowel disease: a review of medical therapy. World J Gastroenterol 2008; 14 : 354–377.
2. Abraham C, Cho JH. Inflammatory Bowel Disease. N Engl J Med 2009; 361 : 2066–2078.
3. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447 : 661–678.
4. Hampe J, Franke A, Rosenstiel P et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39 : 207–211.
5. Massey DC, Parkes M. Common pathways in Crohn’s disease and other inflammatory diseases revealed by genomics. Gut 2007; 56 : 1489–1492.
6. Rioux JD, Xavier RJ, Taylor KD et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39 : 596–604.
7. Van Limbergen J, Wilson DC, Satsanagi J. The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet 2009; 10 : 89–116.
8. Molodecky NA, Kaplan GG. Environmental Risk Factors for Inflammatory Bowel Disease. Gastroenterol Hepatol 2010; 6 : 339–346.
9. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet 2007; 369 : 1627–1640.
10. Rutgeerts P, Goboes K, Peeters M et al. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991; 338 : 771–774.
11. Sartor RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterology 2004; 126 : 1620–1633.
12. Chassaing B, Rolhion N, de Vallé A et al. Crohn disease – associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest 2011; 121 : 966–997.
13. Henderson P, Van Limbergen JE, Schwarze J et al. Function of intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm Bowel Dis 2011; 17 : 382–395.
14. Gaudier E, Hoebler C. Physiological role of mucins in the colonic barrier integrity. Gastroenterol Clin Biol 2006; 30 : 965–974.
15. Ramasundara M, Leach ST, Lemberg DA et al. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol 2009; 24 : 202–208.
16. Wehkamp J, Stange EF, Fellermann K. Defensin-immunology in inflammatory bowel disease. Gastroenterologie Clin Biol 2009; 33 (Suppl 3): 137–144.
17. Kyd JM, Cripps AW. Functional differences between M cells and enterocytes in sampling luminal antigens. Vaccine 2008; 26 : 6221–6224.
18. Ismail AS, Behrendt CL, Hooper LV. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J Immunol 2009; 182 : 3047–3054.
19. Yoshida M, Claypool SM, Wagner JS et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 2004; 20 : 769–778.
20. Marchesi J, Shanahan F. The Normal intestinal microbiota. Curr Opin Infect Dis 2007; 20 : 508–513.
21. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inlammatory bowel disease. Nature 2007; 448 : 427–434.
22. Hugot JP, Chamaillard M, Zouali H et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001; 411 : 599–603.
23. Ogura Y, Bonen DK, Inohara N et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001; 411 : 603–606.
24. Inohara N, Ogura Y, Fontalba A et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 2003; 278 : 5509–5512.
25. Abraham C, Cho JH. Functional consequence of NOD2 (CARD15) mutations. Inflamm Bowel Dis 2006; 12 : 641–650.
26. Tanabe T, Chamaillard M, Oqura Y et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J 2004; 23 : 1587–1597.
27. Lees CW, Sastsanagi J. Genetics of inflammatory bowel disease: implications for disease pathogenesis and natural history. Expert Rev Gastroenterol Hepatol 2009; 3 : 513–534.
28. van Heel DA, Ghosh S, Butler M et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet 2005; 365 : 1794–1796.
29. Wehkamp J, Salzman NH, Porter E et al. Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA 2005; 102 : 18129–18134.
30. Wehkamp J, Wang G, Kübler I et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J Immunol 2007; 179 : 3109–3118.
31. Wehkamp J, Stange EF, Fellermann K. Defensin-immunology in inflammatory bowel disease. Gastroenterologie Clin Biol 2009; 33 (Suppl 3): 137–144.
32. Sartor RB. Does Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease? Gut 2005; 54 : 896–898.
33. Coulombe F, Divangahi M, Veyrier F et al. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med 2009; 206 : 1709–1716.
34. Rahman MK, Midtling EH, Svingen PA et al. The Pathogen Recognition Receptor NOD2 Regulates Human FOXP3+ T Cell Survival. J Immunol 2010; 184 : 7247–7256.
35. Jess T, Riis L, Jespersgaard C et al. Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. Am J Gastroenterol 2005; 100 : 2486–2492.
36. Cho JH, Weaver CT. The genetics of inflammatory bowel disease. Gastroenterology 2007; 133 : 1327–1339.
37. Hradsky O, Lenicek M, Dusatkova P et al. Variants of CARD15, TNFA and PTPN22 and susceptibility to Crohn’s disease in the Czech population: high frequency of the CARD15 1007fs. Tissue Antigens 2008; 71 : 538–547.
38. Lesage S, Zouali H, Cézard JP et al. EPWG-IBD Group; EPIMAD Group; GETAID Group. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 2002; 70 : 845–857.
39. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118 : 229–241.
40. Yamamoto-Furusho JK, Podolsky DK. Innate immunity in inflammatory bowel disease. World J Gastroenterol 2007; 13 : 5577.
41. Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 2000; 68 : 7010–7017.
42. González-Navajas JM, Fine S, Law J et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest 2010; 120 : 570–581.
43. Browning BL, Huebner C, Petermann I et al. Has Toll-like receptor 4 been prematurely dismissed as an inflammatory bowel disease gene? Association study combined with meta-analysis shows strong evidence for association. Am J Gastroenterol 2007; 102 : 2504–2512.
44. Arbour NC, Lorenz E, Schutte BC et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 2000; 25 : 187–191.
45. Ferwerda B, McCall MB, Alonso S et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc Natl Acad Sci USA 2007; 104 : 16645–16650.
46. Watanabe T, Kitani A, Murray PJ et al. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol 2004; 5 : 800–808.
47. Kullberg BJ, Ferwerda G, De Jong DJ et al. Crohn’s disease patients homozygous for the 3020insC NOD2 mutation have a defective NOD2/TLR4 cross-tolerance to intestinal stimuli. Immunology 2008; 123 : 600–605.
48. Schmitd D, Dengjel J, Schoor O et al. Autophagy in innate and adaptive immunity against intracellular pathogens. J Mol Med 2006; 84 : 194–202.
49. Homer CR, Richmond AL, Rebert NA et al. ATG16L1 and NOD2 Interact in an Autophagy-Dependent Antibacterial Pathway Implicated in Crohn’s Disease Pathogenesis. Gastroenterology 2010; 139 : 1630–1641.
50. Kuballa P, Huett A, Rioux JD et al. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 2008; 3: e3391.
51. Saitoh T, Fujita N, Jang MH et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456 : 264–268.
52. Cadwell K, Liu JY, Brown SL et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456 : 259–263.
53. Hussey S, Travassos LH, Jones NL. Autophagy as an emerging dimension of adaptive and innate immunity. Semin Immunol 2009; 21 : 233–241.
54. Parronchi P, Romagnani P, Annunziato F et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol 1997; 150 : 823–832.
55. Duerr RH, Taylor KD, Brant SR et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006; 314 : 1461–1463.
56. Wilson NJ, Boniface K, Chan JR et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 2007; 8 : 950–957.
57. Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009; 58 : 1152–1167.
58. Oppmann B, Lesley R, Blom B et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000; 13 : 715–725.
59. Sandborn WJ, Feagan BG, Fedorak RN et al. Ustekinumab Crohn’s Disease Study Group. A randomized trial of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 2008; 135 : 1130–1141.
60. Monteleone I, Pallone F, Monteleone G. Interleukin-23 and Th17 Cells in the Control of Gut Inflammation. Mediators Inflamm 2009; 2009 : 297645.
61. Andoh A, Zhang Z, Inatomi O et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005; 129 : 969–984.
62. Monteleone G, Caruso R, Fina D et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 2006; 55 : 1774–1780.
63. Liang SC, Tan XY, Luxenberg DP et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203 : 2271–2279.
64. Dambacher J, Beigel F, Zitzmann K et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut 2009; 58 : 1207–1217.
65. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat Immunol 2008; 9 : 641–649.
66. Xu L, Kitani A, Fuss I et al. Cutting edge: regulatory T cells induce CD4+CD25−Foxp3 − T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-β. J Immunol 2007; 178 : 6725–6729.
67. Laurence A, Tato CM, Davidson TS et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26 : 371–381.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2012 Číslo 4
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Akutní infarkt myokardu navozený požitím drogy pervitin
- Akutní intoxikace mědí při suicidiálním pokusu
- Význam biomarkerů NGAL a cystatinu C u kardiovaskulárních onemocnění
- Nové poznatky v patogenezi Crohnovy choroby