
DIAGNOSTIKA A LÉČBA SYSTÉMOVÉ AL AMYLOIDÓZY
Vyšlo v časopise:
Transfuze Hematol. dnes,25, 2019, No. Supplementum 1, p. 37-71.
Kategorie:
Doporučení
A. DOPORUČENÍ PRO DIAGNOSTIKU AL AMYLOIDÓZY
- Systémová AL amyloidóza (ALA) je vzácné hematologické onemocnění patřící mezi monoklonální gamapatie.
- Optimální péče o nemocné s ALA je založena na časné diagnóze s vyhodnocením závažnosti orgánového postižení, účinné terapii zaměřené zejména na snížení produkce amyloidogenních lehkých řetězců a léčbě podpůrné.
- V 12–20 % je asociována s jinou formou monoklonální gamapatie či B-lymfoproliferativního onemocnění.
- Amyloidóza musí být vždy histologicky verifikována spolu s potvrzením AL typu amyloidu.
- Podkladem onemocnění je depozice amyloidu v orgánech a tkáních, což vede k jejich funkčnímu postižení, eventuálně selhání. Pečlivé posouzení orgánového postižení je základním pilířem diagnostiky a stratifikce nemocných.
- Detekce a kvantifikace monoklonálního imunoglobulinu a zejména hladin volných lehkých řetězců patří mezi stěžejní aspekty diagnostiky a sledování nemocných se systémovou ALA.
B. DOPORUČENÍ PRO LÉČBU AL AMYLOIDÓZY
- Principem léčby je eliminace patologického klonu se zastavením produkce amyloidogenních lehkých řetězců, redukcí jejich sérových hladin s cílem dosažení alespoň velmi dobré parciální hematologické remise (úroveň důkazu IIa, stupeň doporučení B).
- Dosažení hematologické léčebné odpovědi je podmínkou pro zlepšení funkce postižených orgánů a dosažení tzv. orgánové léčebné odpovědi (úroveň důkazu IIa, stupeň doporučení B).
- Léčba je volena individuálně s využitím „risk-adapted“ strategie (úroveň důkazu IV, stupeň doporučení C).
- Pouze část nemocných s AL amyloidózou splňující výběrová kritéria je možné bezpečně léčit vysokodávkovanou terapií s podporou autologního štěpu, tito nemocní s nízkou mírou orgánového postižení z této terapie profitují (úroveň důkazu IIa, stupeň doporučení B).
- Léčba kombinací melfalanu a dexamethasonu je možnou variantou pro nemocné, kteří nejsou vhodnými kandidáty vysokodávkované terapie (úroveň důkazu IIa, stupeň doporučení B).
- Kombinovaná schémata s bortezomibem jsou vhodnými léčebnými režimy pro všechny nově diagnostikované nemocné bez ohledu na to, zda jsou či nejsou vhodnými kandidáty vysokodávkované terapie (úroveň důkazu IIa, stupeň doporučení B).
- Kombinovaná schémata s bortezomibem jsou vhodnými léčebnými režimy pro relabující nemocné (úroveň důkazu IIa, stupeň doporučení B).
- Schémata s imunomodulačními látkami jsou indikována u relabujících/refrakterních nemocných (úroveň důkazu IIa, stupeň doporučení B).
- Multioborová spolupráce při péči o nemocné s AL amyloidózou je naprosto nezbytná.
1 METODICKÉ POSTUPY TVORBY DOPORUČENÍ
Předložené doporučení bylo vypracováno na základě medicíny založené na důkazech s cílem popsat všechny důležité oblasti diagnostiky a léčby AL amyloidózy. Úrovně důkazů a stupně doporučení standardně používané v doporučeních jsou uvedeny v tabulce 1.

1.1 Úvod k amyloidózám
Amyloidózy představují heterogenní skupinu onemocnění charakterizovanou přítomností depozit amyloidu, amorfního bílkovinného materiálu uloženého extracelulárně v tkáních. Každý typ amyloidu má svůj specifický fibrilární prekurzorový protein, od něhož se odvíjí i jeho název, např. u familiární transthyretinové amyloidózy jde o molekuly mutovaného trans-thyretinu (ATTR), u AL amyloidózy jde o strukturálně aberantní molekuly lehkého řetězce imunoglobulinu (AL) [Kyle, 1995a; Dispenzieri, 2012]. Podle příčiny produkce amyloidogenního prekurzoru se rozlišují formy získané a hereditární, jež jsou spojeny s mutací genu zodpovědného za syntézu daného prekurzoru. Amyloidóza se vyskytuje ve formě lokalizované s pouze ložiskovou depozicí amyloidu, mnohem častější je ale forma systémová vyznačující se postižením více orgánů a tkání. V současnosti zahrnuje členění založené na typu amyloidových prekurzorů 36 typů, v případě ATTR amyloidózy dalších > 100 variant [Sipe, 2016; Ryšavá, 2013]. Pro potřeby běžné medicínské praxe postačuje jednoduché, klinicky orientované, dělení (tab. 2) [Kyle, 1995a; Falk, 1997; Hassan, 2005].
![Zjednodušená klasifikace nejčastějších typů amyloidóz [Kyle, 1995a; Sipe, 2016]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/ce32ba5bc93036bfde8257aa691f31aa.png)
Z hlediska výskytu je nejčastější systémová AL amyloidóza (70 %), méně častá je její ložisková forma (19 %), zatímco ostatní typy amyloidóz se vyskytují mnohem vzácněji, např. senilní a familiární ve 4 % a AA amyloidóza jen ve 3 % [Kyle, 1992, 1995a]. Historické názvosloví rozpoznávající pouze amyloidózu primární (AL typ) a amyloidózu sekundární (AA typ) se v současnosti již prakticky nepoužívá. Vzhledem k občasnému výskytu a zejména nízké informovanosti lékařů uniká i v současnosti amyloidóza časnému rozpoznání a většinou jsou podchyceny až pokročilé fáze nemoci se závažným, často i nevratným poškozením životně důležitých orgánů. Amyloidóza se vyznačuje sklonem k trvalé progresi a nepříznivé prognóze, neboť všechny typy amyloidózy jsou doposud léčebně jen omezeně ovlivnitelné [Gertz, 2009; Ryšavá, 2013].
2 CHARAKTERISTIKA, VÝSKYT A EPIDEMIOLOGIE SYSTÉMOVÉ AL AMYLOIDÓZY
2.1 Charakteristika
Systémová AL amyloidóza (ALA) je onemocnění vyznačující se pozvolnou monoklonální proliferací plazmocytů v kostní dřeni (plazmocelulární dyskrazie) provázenou lineární tkáňovou depozicí bílkovinného nerozpustného fibrilárního materiálu, tj. amyloidu, jehož prekurzorem jsou lehké řetězce (LŘ) monoklonálního imunoglobulinu (MIg) λ (3krát častěji než κ), nebo jejich fragmenty, jenž jsou prokazatelné v séru a/nebo v moči. Název AL amyloidóza odpovídá charakteru prekurzorového proteinu, tj. A – amyloid, L – lehké řetězce MIg [Bird, 2004; Ščudla, 2009; Gertz, 2009]. Progresivní extracelulární akumulace AL amyloidu v cévní a parenchymové složce životně důležitých orgánů vede k tkáňové dezorganizaci s úbytkem normálních elementů a k progresivní poruše funkce zejména srdce, ledvin, jater, zažívacího traktu, periferního i autonomního nervového systému aj., avšak s výjimkou CNS. Depozita AL amyloidu vyvolávají pouze malou, případně žádnou místní tkáňovou reakci, takže neexistuje přímá souvislost mezi tíží amyloidových depozit a závažností orgánové dysfunkce. Za narušení, případně selhání funkce orgánů, v případě závažného postižení srdce i smrt, je odpovědná i vysoká tkáňová toxicita amyloidogenních lehkých řetězců [Rajkumar, 2007]. Optimální péče o nemocné s ALA je založena na časné diagnóze s vyhodnocením závažnosti orgánového postižení, účinné terapii zaměřené zejména na snížení produkce amyloidogenních LŘ a léčbě podpůrné. Díky prohloubení znalostí charakteristiky ALA dochází v posledních letech i ke zlepšení léčebných výsledků vyznačujících se zlepšením funkce postižených orgánů a prodloužením délky i kvality života nemocných s touto závažnou, život ohrožující, chorobou.
2.2 Výskyt a epidemiologie
AL amyloidóza je nejčastějším typem systémové amyloidózy. Její incidence je v USA odhadována na 5,1–12,8 nemocných na 1 milion obyvatel/rok [Kyle, 1992], v Evropě na 0,8–1,0/100 000 obyvatel/rok [Merlini, 2007]. Systémová ALA postihuje především jedince vyššího věku, v 66 % jde o nemocné mezi 50–70 lety, 17 % je mladších než 50 let a pouze 4 % je mladších 40 let, přičemž poněkud častěji jsou postiženi muži [Kyle,1995b; Bird, 2004].
U systémové ALA se setkáváme s přibližně následujícím zastoupením jednotlivých imunochemických typů MIg: IgG 35 %, IgA 10 %, IgM 5 %, IgD 1 %, ve zbývajících případech jde o přítomnost LŘ λ nebo κ. Ve 12–20 % provází systémová ALA mnohočetný myelom (MM), méně často Waldenströmovu makroglobulinemii (WM) a jen vzácně ostatní lymfoproliferativní stavy.
2.3 Patogeneze
Patogeneze ALA je složitá a doposud nebyla zcela objasněna. Základní příčinou je porucha funkce zvolna proliferujících monoklonálních plazmocytů produkujících klonální lehký řetězec s patologickou konformací (misfolded) mající amyloidogenní potenciál. K tvorbě AL amyloidu dochází v důsledku tvorby makromolekulárních agregátů bílkovinného fibrilárního materiálu za spoluúčasti kodepozice kofaktorů nefibrilárního charakteru. Všechny typy amyloidových depozit obsahují glykoprotein SAP (sérum amyloid P komponenta), který je vysoce odolný proti proteolýze podílející se na rezistenci fibril amyloidu vůči degradaci. Nelze rovněž pominout vliv tkáňového mikroprostředí ovlivňujícího biochemické charakteristiky amyloidogenních LŘ i cytotoxický potenciál solubilních oligomerů [Merlini, 2007]. Dokladem toxicity amyloidových prekurzorů (např. LŘ) je klinické pozorování, že ke zlepšení orgánové funkce (např. poklesu NT-proBNP) dochází již záhy po nasazení terapie, tedy v příliš krátkém intervalu pro uplatnění resorpce fibrilárních depozit [Palladini, 2006].
3 KLINICKÝ OBRAZ SYSTÉMOVÉ AL AMYLOIDÓZY
3.1 Nejčastější projevy nemoci, které jsou indikací k dalšímu vyšetření
Vlastní projevy nemoci jsou velmi pestré a individuálně značně rozdílné, v závislosti na typu a závažnosti postižení orgánů. Pravidelnou součástí klinického obrazu jsou jednak celkové nespecifické projevy, tj. slabost, malátnost, únavnost, snížení chuti k jídlu a úbytek hmotnosti, jednak pestré projevy vyplývající z postižení jednotlivých orgánů a tkání [Bird, 2004; Ščudla, 2009]. Lze je rozdělit na příznaky subjektivní a objektivní, ovšem velmi důležitou manifestací jsou projevy charakteru různých klinických syndromů, jejichž diagnostické řešení může přivést lékaře ke kauzální diagnóze (tab. 3) [Bird, 2004; Gertz, 2005 a 2009; Ščudla, 2009; Ryšavá, 2013].
![Klinické projevy systémové AL amyloidózy [Bird, 2004; Gertz, 2009; Ryšavá, 2013]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/dbc74bff2eaaf06550307f77d34dcfe4.png)
V souboru 868 nemocných se systémovou ALA byl zaznamenán výskyt slabosti v 68 %, edémy v 62 %, pokles hmotnosti v 43 %, dušnost v 40 %, ortostatická hypotenze v 27 %, dysestezie a parestezie v 23 %, dysgeuzie v 18 %, makroglosie v 14 %, kožní purpura v 11 % a průjem v 9 % případů, zatímco projevy přímého postižení CNS jsou pro neprostupnost hematoencefalické bariéry vzácné [Merlini, 2007].
Postižení jednoho orgánu se vyskytuje v 25 %, dvou v 36 %, 3 a více orgánů u 39 % nemocných, zatímco amyloidová nefropatie v 72 %, kardiomyopatie v 63 %, postižení jater v 27 %, periferní neuropatie v 19 %, postižení autonomního nervstva v 16 %, postižení měkkých tkání v 12 % a amyloidová kožní purpura v 11 % [Merlini, 2007]. Bylo zjištěno, že nemocní se systémovou ALA a přítomností MIg typu IgM mají oproti IgG typu poněkud odlišný klinický obraz: u poloviny nemocných jsou nejčastěji postiženy ledviny a měkké tkáně, častěji, a to v 21 %, jsou postiženy lymfatické uzliny a respirační aparát, naopak méně často, a to v 36 %, je postiženo srdce. U více než poloviny nemocných jsou při diagnóze známky postižení více než dvou orgánů a v 73 % infiltrace kostní dřeně lymfoplazmocytárními elementy [Wechalekar, 2008]. Nemocní s přítomností IgD typu M-proteinu se pak vyznačují nízkým výskytem postižení ledvin a srdce [Gertz, 2012].
3.2 Nejvýznamnější klinické projevy AL amyloidózy
3.2.1 Amyloidóza ledvin
Postižení funkce ledvin, zejména jejich glomerulárních funkcí, je nejčastějším ale málo specifickým projevem a nezávisí pouze na přítomnosti amyloidových depozit, ale i na stupni intersticiální fibrózy a tubulární atrofie [Bohle, 1993]. Klíčovým projevem vedoucím obvykle k odhalení ALA je výrazná proteinurie charakteru nefrotického syndromu (NS) vyznačujícího se navíc hypoalbuminemií, hyperhydratací, generalizovanými otoky včetně ascitu, pleurálního a perikardiálního výpotku, tj. případnou anasarkou s enormním nárůstem hmotnosti o 15–20 kg [Ryšavá, 2013]. Perzistentní NS se současným selháním ledvin a se zvětšením jejich velikosti je pro amyloidovou nefropatii velmi sugestivní. Dokud nedojde k infiltraci intrarenálních cévních struktur amyloidem, není přítomna arteriální hypertenze, která navíc nepatří do klinického obrazu amyloidové nefropatie [Dember, 2010]. Celkový katabolismus s negativní dusíkovou bilancí vede ke ztrátě svalové hmoty spojené s hypotonií a atrofií svalstva. NS se může v důsledku ztrát imunoglobulinů močí komplikovat zvýšeným výskytem infekcí, ale i tromboembolickou nemocí s trombózou renálních žil. Hyperlipoproteinemie provázená xantelasmaty víček a/nebo oblasti extenzorů vede k projevům akcelerované aterosklerózy, ztráta transferinu močí bývá jednou z příčin refrakterní anemie. Jen zřídka dochází k manifestaci Fanconiho syndromu (postižení proximálního tubulu), zatímco nefrogenní diabetes insipidus (postižení distálního tubulu) je spíše výrazem koincidence s MM [Ryšavá, 2013].
3.2.2 Amyloidóza srdce
Postižení srdce u systémové ALA je velmi časté a při diagnóze bývá přítomno u více než 2/3 nemocných. Při autopsii se nacházejí myokardiální depozita AL amyloidu prakticky vždy, samotné izolované postižení myokardu je ale vzácné, pouze v 5 %. Depozita amyloidu vedou k progresivnímu zesílení stěn obou komor, což porušuje plnění srdce v průběhu diastoly, vzniká tedy obraz restriktivní, resp. hypertrofické kardiomyopatie. Infiltrace chlopenního a závěsného aparátu amyloidem může navodit chlopenní vady charakteru mitrální a trikuspidální insuficience, vzácně lze pozorovat perikarditidu, zcela raritně byla popsána ruptura stěny pravé komory. Manifestní srdeční selhání je vedoucím příznakem u 25 % nemocných, přičemž v popředí klinického obrazu jsou zejména projevy pravostranné srdeční slabosti nebo známky sníženého srdečního výdeje vedoucího k poklesu krevního tlaku, což spolu s projevy autonomní neuropatie vede k ortostatické a/nebo chronické hypotenzi a k častým synkopálním stavům. Projevy iniciálně diastolické, posléze i systolické dysfunkce vedou nakonec k rozvoji globálního srdečního selhání s nízkou tolerancí tělesné zátěže, dušností, výskytu periferních otoků a nakonec i ascitu, pleurálnímu výpotku a anasarky. Častým projevem jsou i palpitace, synkopy v rámci rytmických poruch, např. supraventrikulární arytmie, syndrom chorého sinu a AV blokády. Nepříliš časté jsou anginózní bolesti při postižení drobných koronárních tepen depozity amyloidu a aterosklerózou. I v současnosti platí, že postižení srdce amyloidem je nejvýznamnější prognostický faktor, jenž má vliv na volbu léčebné strategie a na délku celkového přežití [Kyle, 1986]. Nejčastější příčinou smrti u systémové AL amyloidózy je pokročilé, léčebně refrakterní, srdeční selhání, náhlá smrt v důsledku elektromechanické disociace nebo maligní komorová arytmie [Pika, 2008; Fikrle, 2013; Paleček, 2015; Quarta 2012; Rajkumar, 2013].
3.2.3 Periferní a autonomní neuropatie
Periferní neuropatie se projevuje širokou škálou příznaků, přičemž postižení hlavových nervů je vzácné. Dostavuje se obvykle záhy v oblasti DKK a se značným předstihem před vlastním rozpoznáním ALA. Je zpravidla axonální, charakteru distální smíšené, symetrické, periferní neuropatie projevující se přítomností dysestezie, parestezií s progresivní ztrátou citlivosti, trnutím a svalovou slabostí [Gertz, 2009]. Senzitivní neuropatie je obvykle symetrická, někdy značně bolestivá, zatímco motorická neuropatie je podstatně vzácnější. Do klinického obrazu patří i „syndrom neklidných nohou“. Častým projevem je syndrom karpálního tunelu, nezřídka předcházející ostatní projevy AL amyloidózy o několik let, vznikající v důsledku depozit amyloidu v oblasti vazů karpálního tunelu. Přítomný edém a útlak n. medianus se projevuje především nočními paresteziemi v oblasti dlaní, palce, ukazováku a prostředníčku s atrofií svalů v oblasti tenaru a hypotenaru [Kyle, 1995a].
Autonomní neuropatie je často v popředí klinického obrazu, neboť se vedle postižení srdce a hypokortikalismu podílí na rozvoji posturální hypotenze, kdy systolická hodnota TK se snižuje v ortostáze v odstupu 3–5 minut polohy ve stoje o > 20 mm Hg, obvykle na < 90 mm Hg. Projevuje se rovněž poruchou motility GIT s časným falešným pocitem sytosti a dysgeuzií, u mužů erektilní i ejakulační dysfunkcí, dysfunkcí močového měchýře s obtížným vyprazdňováním, ale i anhidrózou.
3.2.4 Amyloidóza GIT a jater
Gastrointestinální projevy vznikají v důsledku depozice amyloidu ve stěně GIT a v důsledku poruchy vegetativního systému. Nápadným, do jisté míry i příznačným, projevem AL amyloidózy je makroglosie způsobená imbibicí jazyka amyloidem. Vzhledem k dominantnímu postižení kořene jazyka může vést nejen k poruše polykání, ale i k výraznému zúžení dýchacích cest. Dyspeptický syndrom dostavující se v důsledku gastroparézy bývá spojen s pocity předčasné sytosti, nauzeou a zvracením, postižení nižších etáží se vyznačuje bolestí břicha, obstipací (pseudoobstrukce), ale i obrazem explozivního postprandiálního průjmu s malabsorpcí a s úbytkem hmotnosti. V rámci amyloidózy GIT byla popsána i perforace střeva, střevní infarzace a výrazné krvácení z distálních oblastí zažívací trubice jako důsledek zvýšené fragility cév. Zvětšení jater s pozitivitou jaterních testů, zejména jaterní frakce alkalické fosfatázy, a portální hypertenze s ascitem a splenomegalií se vyskytují asi u 1/4 nemocných [Bird, 2004].
3.2.5 Postižení ostatních orgánů a tkání
Postižení dýchacích cest a plic se projevuje kašlem, dušností a hemoptýzou. Infiltrace stěny laryngu vede k obstrukci horních dýchacích cest, inspirační dušnosti a postižení hlasivek s dysfonií. Vzácně lze zachytit ložiskové nodulární postižení plic (coin lession, resp. amyloidom) a/nebo amyloidovou infiltraci tracheobronchiálního stromu, vedoucí ke známkám obstrukce, zatímco postižení pleury se vyznačuje přítomností torpidního pleurálního výpotku.
Poměrně časté jsou i poruchy hemostázy, asi u 1/4 nemocných. Nejčastěji je přítomna vaskulární kožní purpura, např. periorbitální nebo v oblasti hřbetu rukou a předloktí, tzv. „syndrom červených očí“ v důsledku postižení cév spojivkového vaku s vývinem subkonjunktivální sufuze nebo slizniční krvácení v různých oblastech jako důsledek zvýšené fragility cév při subendoteliální depozici amyloidu. Na projevech hemoragické diatézy se může podílet i snížení počtu krevních destiček při splenomegalii a významné snížení hladiny faktoru X vedoucí k život ohrožujícímu krvácení po jaterní nebo renální biopsii [Bird, 2004; Mumford, 2000]. Makroskopická hematurie může být výsledkem výše uvedených poruch hemostázy, ale i důsledkem infiltrace vývodných močových cest amyloidem.
Infiltrace nadledvin amyloidem se projevuje adrenální insuficiencí s obrazem Addisonovy choroby, inbibice štítné žlázy hypotyreózou. K vzácným projevům ALA patří umístění amyloidových depozit v oblasti očních spojivek, sklér, víček a orbity. Infiltrace lymfatických uzlin se manifestuje zvětšením uzlin, amyloidová myopatie svalovou slabostí v důsledku poškození a imbibice příčně pruhovaného svalstva amyloidem, depozita amyloidu v synovii a přilehlých kloubních strukturách obrazem neerozivní symetrické artropatie s predilekčním postižením ramenních, zápěstních, drobných ručních a kolenních kloubů. Postižení kůže se manifestuje jejím ztluštěním, případně přítomností papul a plaků, vzácně obrazem napodobujícím sklerodermii, postižení nehtů se ohlašuje onychodystrofií [Kyriakides, 2002].
4 VYŠETŘOVACÍ TECHNIKY U SYSTÉMOVÉ AL AMYLOIDÓZY
4.1 Přehled vyšetřovacích metod
Vzhledem k neuspokojivým výsledkům léčby pokročilé fáze systémové ALA je naléhavým požadavkem rozpoznat onemocnění již v jeho počáteční fázi. Klíčovým předpokladem správné diagnózy je požadavek, aby lékaři v klinické praxi na toto onemocnění mysleli. K podezření na diagnózu ALA by měla vést zejména přítomnost nefrotického syndromu s případnou renální insuficiencí, nedilatační kardiomyopatie, nejasné periferní a/nebo autonomní neuropatie a ostatních klinických syndromů resp. mnohotvárných subjektivních příznaků a objektivních příznaků (viz tab. 3, kap. 3 Klinický obraz systémové AL amyloidózy). Za těchto okolností je povinností lékaře pátrat po přítomnosti MIg standardní a imunofixační elektroforézou séra, resp. zvýšených hladinách VLŘ včetně patologie poměru κ/λ a v případě jejich přítomnosti odeslat nemocného k dovyšetření na nejbližší hematologické oddělení [Bird, 2004; Ščudla, 2009; Gertz, 2009, 2010, 2016]. Včasné rozpoznání systémové ALA vyžaduje vedle dobré znalosti nemoci i notnou dávku diagnostické invence. Přehled základních i výběrově indikovaných vyšetření při podezření na systémovou ALA je uveden v tabulce 4 [Dispenzieri, 2012, Gillmore, 2014], vlastní diagnostický algoritmus je shrnut v tabulce 5 (viz kap. 5 Diagnostická kritéria a postupy u systémové AL amyloidózy).
![Přehled vyšetření nezbytných pro diagnózu a určení stadia AL amyloidózy (volně podle [Dispenzieri, 2012, Gillmore, 2014])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/d5ee5ed98c14766fe298d17c3a49d25c.png)
![Diagnostická kritéria systémové AL amyloidózy dle IMWG (upraveno podle [Rajkumar, 2011; Gertz, 2016])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/484a201832f6d92add818a4303d931ba.png)
4.2 Standardní techniky v diagnostice systémové AL amyloidózy
4.2.1 Diagnostika amyloidu z pohledu patologa
Amyloidem nazýváme substanci tvořenou amyloidotvorným fibriláním proteinem a dalšími přídatnými složkami (serový amyloidový protein P, lipoproteiny, glykosaminoglykany, proteoglykany) [Pepys, 2006; Vrana, 2009; Vrana, 2014; D’Souza, 2013]. Amyloidotvorný fibrilární protein nabývá vzhledu rigidních nevětvících se fibril průměru přibližně 10 nm, které jsou nerozpustné a jsou výsledkem změn v uspořádání proteinových molekul.
V běžné morfologické praxi se s amyloidem setkáváme v základním a speciálním histologickém barvení, v imunohistochemii, imunofluorescenci, méně často se využívá elektronové mikroskopie. Mezi další užitečné metody sloužící k identifikaci amyloidových fibril patří Western blotting, sekvenování aminových kyselin a imunoelektronová mikroskopie [Sipe, 2016].
Metody rozdělujeme na detekční – pro zjištění přítomnosti amyloidu (speciální histologické barvení, elektronová mikroskopie) a typizační – pro určení konkrétního typu amyloidotvorného proteinu (imunohistochemie, imunofluorescence, imunoelektronová mikroskopie, proteomická analýza, …). Zájemce o tuto problematiku odkazujeme na uvedenou literaturu a také na text schválený Společností českých patologů ČLS JEP (Minimální požadavky na diagnostický postup pro detekci amyloidu v rámci systémových amyloidóz).
Histologické vyšetření v základním a speciálním barvení
Depozita amyloidu se v základním barvení histologických preparátů (hematoxylin-eosin) znázorní nespecificky jako amorfní eozinofilní, tj. světle červený materiál. Pro specifické znázornění amyloidu existují speciální barvicí metody, přičemž pro diagnózu nepodkročitelným standardem je celosvětově používané barvení Kongo červení. V jeho výsledku poskytují depozita amyloidu oranžové až červené zbarvení, a to bez závislosti na typu amyloidu [Vacek, 1988]. Při vyšetření polarizačním mikroskopem pozorujeme charakteristické jevy, které dává konžská červeň ve vazbě na amyloid. Tradičně se označují jako dvojlom (birefringence) a dichroismus apple-green barevného charakteru, avšak jedná se o složité optické jevy komplexního charakteru a výsledné zbarvení může nabývat i jiných odstínů, např. žluté, oranžové či modré [Gertz, 2010; Howie, 2008; Sipe 2016].
Z ostatních speciálních histologických barvicích metod lze použít saturnovou červeň, thioflavin, methylvioleť apod. Lze jen doporučit provádět pro detekci amyloidu spolu s Kongo červení ještě alespoň jednu další speciální barvicí metodu. Speciální barvicí metody dávají dobrý výsledek při klasickém zpracování histologického materiálu – fixace formalinem a zalití do parafinu. Obdobně jako u ostatních barvicích metod dosáhneme kvalitnějšího výsledku barvením zmrazených řezů nefixované tkáně.
Imunohistochemické vyšetření
V současné diagnostice amyloidu zůstává imunohistochemie (IHC) nejvíce dostupnou rutinně používanou metodou, která umožňuje provést vyšetření z materiálu fixovaného formalinem a zalitého do parafinu (FFPE) s příznivými náklady a bez nutnosti pořízení specializovaného přístrojového vybavení.
Do běžné praxe patří vyšetření protilátkami proti κ a λ lehkých řetězců imunoglobulinů, transthyretinu a amyloidu A, přičemž spektrum komerčně dostupných primárních protilátek i proti vzácným a hereditárním formám amyloidózy se stále rozšiřuje. Depozita všech typů amyloidu obsahují konstantně příměs i jiných molekul, než je samotný amyloidogenní protein, což lze imunohistochemickým průkazem využít v diagnostice (zejm. protilátka proti sérovému amyloidovému proteinu P).
Senzitivita IHC vyšetření amyloidu (zejména AL typu) je ztížena několika okolnostmi. Kromě faktorů preanalytické fáze vyšetření (odběr materiálu, způsob a délka fixace apod.) hraje významnou roli samotný proces tvorby amyloidu (konformační změny s fragmentací lehkých řetězců a maskování antigenu při formování amyloidových fibril), dále přirozená heterogenita lehkých řetězců plynoucí z jejich variabilních domén, mutace amyloidotvorného proteinu a v neposlední řadě příměs séra. Také rozpoznávání epitopů jednotlivými protilátkami může být odlišné. Výše uvedené vede ke skutečnosti, že závěry studií, které zkoumají spolehlivost IHC vyšetření v určování subtypů amyloidózy, se často značně liší. V současné době je možné využít více polyklonálních i monoklonálních protilátek proti epitopům κ i λ lehkých řetězců. Aplikace více různých protilátek proti lehkým řetězcům imunoglobulinů zvyšuje validitu vyšetření AL amyloidu. Dle jedné ze studií, použití čtyř protilátek anti-λ v kombinaci se souvisejícími klinickými a laboratorními vyšetřeními přináší senzitivitu 94 % a specificitu 100 % [Schönland, 2012].
Fluorescenční vyšetření
Vyšetření amyloidu pomocí fluorescenčního mikroskopu lze provést pomocí přímé vazby fluorochromu na amyloidová depozita, což nastává v případě barvení parafinového řezu konžskou červení, která sama o sobě je fluorochromem. Ve srovnání s vyšetřením pouze polarizačním mikroskopem vykazuje fluorescenční vyšetření vyšší senzitivitu, zejména v případech málo objemných depozit amyloidu [Marcus, 2012]. Metodou volby je však vyšetření s využitím komerčně dostupných protilátek, které jsou značeny fluorochromem. Pro imunofluorescenci je nutné použít řezy připravené ze zmrazené nefixované tkáně, což vyžaduje přizpůsobit průběh odběrové fáze, neboť transport do laboratoře musí proběhnout bezprostředně po odběru a za odlišných podmínek oproti tkáním standardně zpracovávaným metodou FFPE. V tomto režimu jsou rutinně vyšetřovány vzorky z biopsie ledvin, na některých pracovištích také endomyokardiální biopsie. Imunofluorescenční vyšetření vykazuje vyšší diagnostickou úspěšnost (v různých studiích 67–88 %) [Collins, 2009] ve srovnání s imunohistochemií, nevýhodou a určitou limitací však stále zůstává nutnost použít nefixovaný mražený materiál, neboť amyloid je velmi často náhodným nálezem. V případě vysoké klinické suspekce na amyloid je již při prvním odběru necílené biopsie vhodné poslat do laboratoře část odebraného materiálu také na imunofluorescenční vyšetření. Novou možností vyšetření je metoda, která využívá natrávení parafinových fixovaných vzorků proteázami (protease digested), které lze následně vyšetřit imunofluorescenčně na průkaz depozice lehkých řetězců. Lze tak dodatečně provést imunofluorescenční vyšetření z fixovaných vzorků anebo zopakovat vyšetření z materiálu, kde přímá imunofluorescence nebyla přesvědčivá (pro průkaz κ řetězců se tato metoda zdá i přesnější) [Nasr 2006].
Elektronová mikroskopie
Charakteristickou fibrilární strukturu amyloidu lze prokázat vyšetřením elektronovým mikroskopem, který znázorní nevětvené splétající se mikrofibrily šíře 7,5–10 nm tvořící nepravidelnou síť. Stavba jednotlivých fibril se liší v závislosti na amyloidogenním proteinu. Vysoce specializovaná pracoviště mají k dispozici imunoelektronovou mikroskopii, která spojuje výhody vysokého rozlišení elektronového mikroskopu se specificitou reakce antigen-protilátka a umožňuje ověřit kolokalizaci imunohistochemické reakce a amyloidové fibrily [Steusloff, 2000; Arbustini, 2002].
4.2.2 Průkaz MIg a VLŘ v séru a/nebo moči
Detekce a kvantifikace MIg a hladin volných lehkých řetězců (VLŘ, FLC) včetně abnormálního poměru κ/λ patří mezi stěžejní aspekty diagnostiky a sledování nemocných se systémovou ALA. Standardní elektroforéza bílkovin séra (SPE) je stále základní, ovšem málo citlivou metodou odhalující přítomnost MIg pouze u ~ 50 % AL amyloidóz, přičemž hodnota M-proteinu nebývá vysoká (u ~ 70 % jedinců je < 20 g/l) [Gertz, 2009; Bradwell, 2015]. Proto základním vyšetřením je imunofixační elektroforéza (IFE) odhalující přítomnost MIg nebo LŘ v 71, resp. 84 %, nikoliv tedy u všech nemocných. Předností IFE je schopnost detekce kteréhokoliv imunochemického typu MIg, a to i v případě, že se vyskytuje v séru a/nebo v moči v malém množství. Nevýhodou IFE je nemožnost monitorace průběhu léčby, neboť neposkytuje kvantitativní výsledky [Gertz, 2009; Bradwell, 2010; Maisnar, 2012]. Většina nemocných se systémovou ALA se vyznačuje nízkou hladinou nebo chyběním intaktního MIg, charakteristické je zvýšení hladiny VLŘ λ nebo κ [Bradwell, 2015]. V současnosti je proto již naprosto standardním a neopominutelným postupem kvantitativní stanovení sérové hladiny VLŘ včetně indexu κ/λ s pomocí nefelometrické nebo turbidimetrické techniky (FreeliteTM), dosahující při kombinaci se SPE a IFE 98–99% senzitivity [Gertz, 2009; Bradwell, 2010; Katzmann, 2005; Vávrová, 2012]. Zvýšená hladina VLŘ v séru spolu s patologií indexu κ/λ se vyskytuje u ALA v 86–98 % a koreluje s náloží amyloidu [Lachmann, 2003; Comenzo, 2012]. Sledování pohybu sérových hladin VLŘ u ALA je metodou volby při monitorování a hodnocení hloubky léčebné odezvy, současně hodnota při diagnóze je velmi důležitým prognostickým faktorem s úzkým vztahem k celkovému přežití [Palladini, 2010b]. Rozdíl mezi hladinami postiženého a alternativního řetězce (dFLC – diference) v době diagnózy ≥ 50 mg/l definuje měřitelné onemocnění pomocí hladin VLŘ. Avšak přibližně u 10–15 % nemocných nacházíme pouze minimálně abnormální hladiny VLŘ, pro část nemocných lze užít modifikovaná kritéria léčebné odpovědi, nicméně u dalších nelze stanovení VLŘ pro monitorování užít [Palladini, 2010b]. U malé části nemocných, zejména s těžkým nefrotickým syndromem, bývají často nacházeny nízké hladiny obou typů lehkých řetězců z důvodu úniku do moči, u části těchto nemocných je možné detekovat a kvantifikovat MIg či Benceovu-Jonesovu bílkovinu analýzou moči. Koncentrace VLŘ v séru je jedním ze tří základních kritérií recentního revidovaného stratifikačního systému AL amyloidózy (viz kap. 7.2 Klinická stratifikace).
4.2.3 Zobrazovací techniky u systémové AL amyloidózy
Využití zobrazovacích metod u ALA sleduje dva hlavní aspekty, tj. morfologické a funkční posouzení orgánového postižení amyloidem (zejména myokardu). K detekci amyloidových depozit v organismu lze využít i scintigrafické vyšetření.
Radiografické vyšetření (RTG). Radiografické vyšetření v diagnostickém algoritmu samotné amyloidózy nemá větší diagnostický přínos a dominantně je využíváno k vyloučení osteolytického postižení a tím i asociace systémové ALA s MM (viz kap. 8 Systémová AL amyloidóza a MM). Pomocí RTG hrudníku lze prokázat rozšíření srdečního stínu s městnáním v malém oběhu, pleurální výpotek a vzácně postižení plic v rámci ALA [Georgiades, 2004].
Počítačová tomografie (CT). CT vyšetření má u systémové amyloidózy limitovaný význam zejména z důvodu nízké specificity a hlavní využití představují ložiskové formy onemocnění zejména v oblasti orbity, nazofaryngu či laryngu. Dominantně je však CT vyšetření využíváno k detekci amyloidózy v oblasti tracheobronchiálního stromu (fokální nebo difuzní submukózní depozita zobrazující se jako noduly, plaky či cirkulární ztluštění stěny) a k posouzení postižení plicního parenchymu (nodulární parenchymatózní amyloidóza nebo difuzní alveolární septální amyloidóza). CT vyšetření břicha je přínosné v diagnostice a diferenciální diagnostice jaterního postižení, přičemž diagnostická kritéria vyžadují hepatomegalii > 15 cm.
Ultrazvukové vyšetření (UZ). Doménou ultrazvukové diagnostiky u ALA je echokardiografické (ECHO) posouzení srdečního postižení. ECHO je v současnosti stěžejní metodou pro diagnostiku a sledování srdečního postižení při amyloidóze. Kritériem postižení je koncentrická hypertrofie myokardu levé komory s diastolickou šíří interventrikulárního septa > 12 mm při absenci hypertenze nebo jiné příčiny hypertrofie myokardu [Gertz, 2005]. Dalšími ECHO nálezy jsou diastolická dysfunkce s restriktivním plněním levé komory, snížená ejekční frakce, ztráta longitudinální kontrakční schopnosti levé komory, dilatace levé síně a přítomnost chlopenních vad. Typickým nálezem je sonografický obraz jemně granulovaného myokardu (granular sparkling) [Fikrle, 2013; Esplin, 2013]. ECHO je spolu se stanovením kardiálních biomarkerů stěžejní metodou v hodnocení orgánové léčebné odpovědi, resp. progrese onemocnění. Vybraná pracoviště dnes využívají nových echokardiografických modalit k detailnímu posouzení srdečního postižení, tj. tkáňový Doppler, strain, speckle tracking [Piper, 2010; Koyama, 2010].
Magnetická rezonance (MR). Stejně jako v případě UZ vyšetření je magnetické rezonance využíváno zejména v hodnocení srdečního postižení. Vyšetření srdce magnetickou rezonancí umožňuje morfologické a funkční zobrazení, navíc při užití kontrastního vyšetření gadoliniem dovoluje posouzení rozsahu imbibice myokardu amyloidovými masami. Fenomén pozdního sycení (delayed enhancement) ve své difuzní subendokardiální či méně časté transmurální formě zobrazení je nález charakteristický pro postižení myokardu amyloidem [Maceira, 2005; Hosch, 2008; Ruberg, 2009; Fikrle, 2016]. Vyšetření je vysoce senzitivní a specifické, nicméně pro využití ke sledování nemocných a posuzování léčebné odpovědi nejsou zatím k dispozici dostatečná data. MR je užitečná i v posuzování ložiskové amyloidózy v oblasti laryngu, nazofaryngu, míchy či urogenitálního traktu [Georgiades, 2004].
Radioscintigrafie s využitím značeného 123I-SAP je neinvazivní, kvantitativní a velmi citlivou metodou (u AL a AA amyloidózy 90% senzitivita) pro celotělovou detekci orgánové distribuce amyloidu s dobrou vizualizací depozit v ledvinách, játrech, kostech, slezině a nadledvinkách, a tím i vhodnou technikou k monitorování výsledků léčby. 123I-SAP scintigrafie umožňuje vizualizaci amyloidových depozit i v orgánech bez známek klinického postižení a v případě nemožnosti biopsie [Hawkins, 2002; Palladini, 2010a]. Celosvětově nízká dostupnost 123I-SAP je podmíněna potenciálním rizikem přenosu infekce, neboť SAP se připravuje z krve dárců. V České republice není tato metoda zavedena.
V současnosti je na některých pracovištích užíváno 99mTc-3,3-diphosphono-1-2-propanodicarboxylic acid (DPD) scintigrafie. 99mTc-DPD je selektivně vychytáván v myokardu s depozitity TTR amyloidu, což je výhodné nejen pro diagnostiku a posuzování postižení srdce u hereditární a senilní ATTR amyloidózy, ale i pro diferenciální diagnostiku vůči ALA, u které akumulace radiofarmaka chybí či je velmi nízká [Rapezzi, 2011; Perugini, 2005].
4.3 Speciální diagnostické techniky u systémové AL amyloidózy
4.3.1 DNA analýza
Analýza DNA je důležitým diagnostickým krokem zvláště pro stanovení hereditárních amyloidóz, což jsou onemocnění s geneticky podmíněnou změnou struktury proteinu. Pro genomické vyšetření pacientů s podezřením na hereditární formu amyloidózy je užívána detekce nukleových kyselin pomocí metody PCR (polymerázová řetězová reakce) s následným zjišťováním pořadí nukleových kyselin v polynukleotidovém řetězci (tzv. Sangerovo sekvenování). Cílem je zjistit možnou záměnu nukleotidu ve sledovaném genu, která následně může způsobit záměnu aminokyseliny v polypeptidovém řetězci, patologickou konformaci proteinu a tím i jeho dysfunkci [Sipe, 2016]. Jde o relativně jednoduchou metodiku zavedenou do praxe nejméně na dvou pracovištích v České republice (Praha, Ostrava). V současné době již existuje zavedený a validovaný panel pro diagnostiku hereditárních amyloidóz pomocí sekvenování nové generace (NGS). Tento panel by v budoucnu měl nahradit vyšetřování pomocí Sangerova sekvenování a snížit finanční i časovou náročnost vyšetření [Chyra, 2018].
4.3.2 Hmotnostní spektrometrie
Hmotnostně spektrometrická analýza amyloidních depozit. Hmotnostně spektrometrická proteomická analýza je založená na přímé identifikaci proteinů ve vzorku [Mann, 2001]. Před samotnou analýzou jsou proteiny extrahovány při typizaci amyloidních depozit z tkáňového vzorku a následně jsou proteiny enzymaticky štěpeny na peptidy. Vzniklá peptidová směs je separována pomocí kapalinové chromatografie, kdy se využívá rozdílné hydrofobicity jednotlivých peptidů. Během analýzy jsou jednotlivé peptidy postupně eluovány z kolony, ionizovány a analyzovány na hmotnostním spektrometru. Při analýze na hmotnostním spektrometru je změřena přesná hmota peptidu a také je u něj změřeno fragmentační spektrum. Z těchto informací je možné vyčíst sekvenční informaci o peptidu, tj., z jakých aminokyselin je peptid složen. Pokud je sekvenční informace peptidu unikátní, je identifikován unikátní protein. Z naměřených intenzit signálů pro jednotlivé peptidy je možné získat kvantitativní informaci (abundanci) o jednotlivých proteinech ve vzorku. Výsledkem hmotnostně spektrometrické proteomické analýzy vzorku je list identifikovaných proteinů s jejich kvantitativní informací [Mann, 2001]. Při analýze amyloidních depozit je hledán v listu identifikovaných proteinů nejvíce abundantní amyloidní protein, který určuje typ amyloidu [Dogan, 2017].
Vzorky materiálu fixovaného formalinem a zalitého do parafinu (FFPE vzorky biopsie postiženého orgánu). Hmotnostně spektrometrická proteomická metoda využívá toho, že v dnešní době jsme schopni efektivně extrahovat proteiny ze vzorků materiálu fixovaného formalinem a zalitého do parafinu (FFPE) [Dogan, 2017]. Navíc zavedením laserové mikrodisekce je možné získat Kongo červení obarvená amyloidní depozita, což vede ve svém důsledku k zesílení signálu a významné redukci pozadí ze samotné tkáně. Metoda vyžaduje velmi malé množství (~ 1 nl) amyloidních depozit, které jsou dostupné v bioptických FFPE vzorcích [Theis, 2013]. Identicky i vzorky z biopsie podkožní tukové tkáně můžou sloužit k vyhledávání systémové amyloidózy [Dogan, 2017; Vrana, 2014].
4.3.3 Vyšetření klonality plazmocytů pomocí multiparametrické průtokové cytometrie
Vzhledem k tomu, že pro ALA je typické nízké zastoupení plazmocytů (PC), teprve detekce a identifikace dostatečného počtu PC na základě exprese povrchových znaků CD38 a CD138 umožňuje jejich detailní analýzu. Na rozdíl od MM, kde již kostní dřeň (KD) bývá infiltrována vesměs klonálními PC, u ALA, podobně jako u monoklonální gamapatie nejasného významu (MGUS), se v KD vyskytují v různých poměrech patologické i fyziologické PC. V takovýchto případech je identifikace a enumerace patologických PC pouze na základě stanovení „normality“ dle povrchového fenotypu s využitím znaků CD19 a CD56 (normální N-PC: CD19+CD56 – vs. abnormální A-PC: CD19-CD56+/–) často nepřesná [Kovářová, 2011]. Přesto však bylo zjištěno, že stanovení infiltrace kostní dřeně PC pomocí průtokové cytometrie a procentuální zastoupení počtu normálních a abnormálních PC mohou sloužit jako prognostické parametry ovlivňující celkové přežití pacientů s ALA [Paiva, 2011; Jelinek, 2017]. Pro spolehlivé stanovení přítomnosti a počtu klonálních PC je však nezbytné stanovení intracelulární „klonality“ s využitím kombinace minimálně šesti znaků (CD38, CD138, CD19, CD56, cκ a cλ), ještě lépe však v osmibarevné kombinaci (CD38, CD138, CD19, CD56, CD45, CD27, cκ a cλ) umožňující přesnější odlišení polyklonáních (N-PC) od klonálních (A-PC) plazmocytů. Analýza klonality PC u ALA je pak nezbytným nástrojem pro zhodnocení léčebné odpovědi, kdy dosažení hematologické kompletní remise je nutným předpokladem dosažení orgánové léčebné odpovědi [Adam, 2012b].
5 DIAGNOSTICKÁ KRITÉRIA A POSTUPY U SYSTÉMOVÉ AL AMYLOIDÓZY
5.1 Diagnostická kritéria
Časná diagnóza systémové ALA je obtížná, poněvadž neexistuje žádný samostatný laboratorní ukazatel ani zobrazovací technika, které by diagnózu nemoci samy o sobě umožnily [Gertz, 2011]. Správná diagnóza systémové ALA se proto opírá o splnění souboru diagnostických kritérií sestavených v roce 2011 IMWG (International Myeloma Working Group) v tabelární formě (tab. 5).
Z předložených kritérií je zřejmé, že diagnóza systémové ALA vyžaduje splnění všech čtyř základních kritérií nemoci:
- Jednoznačný průkaz, že k postižení některého z orgánů, tj. ledvin, srdce, GIT včetně jater, nervové tkáně a vzácněji svalů, kůže aj., došlo v důsledku amyloidózy, nikoliv v rámci jiného onemocnění (např. diabetes mellitus, ateroskleróza aj.).
- V klinické praxi se k detekci amyloidu používá standardně současná aspirační biopsie podkožního tuku (pozitivita 60–80 %) a biopsie kostní dřeně (50–65 %), při jejich negativitě i slinné žlázy (asi 15% pozitivita při negativitě amyloidu v podkožním tuku), případně a to i v souladu s klinickým obrazem (např. makroglosie) jazyka, rekta a gingivy. Při diagnostickém neúspěchu necíleného odběru se přistupuje k cílené biopsii dominantně postiženého orgánu, tj. ledviny, myokardu (endomyokardiální biopsie), jater, zažívací trubice, ojediněle i n. suralis, kosterního svalstva, uzliny aj., vyznačující se 80–100% výtěžností s následující hierarchií pozitivity: endomyokardiální biopsie (100 %), biopsie jater (97 %), ledvin (94 %), kůže (90 %), n. suralis (86 %), tenkého střeva (83 %), tkáně z oblasti karpálního tunelu (82 %), biopsie podkožního tuku s hodnocením tří vzorků (80 %), biopsie rekta (75 %) a trepanobiopsie kostní dřeně (56 %). (kap. 4.2.1 Diagnostika amyloidu z pohledu patologa) [Gertz, 1991, 2009, 2016].
- Vzhledem k jisté nespolehlivosti imunohistochemické identifikace AL typu amyloidu používaného ve standardní klinické praxi (v 50 % ztráta antigenních epitopů v průběhu fibrilogeneze) se v současnosti mnohdy užívá kombinace technik, preferována je hmotnostní spektrometrie, na řadě pracovišť považovaná za „zlatý standard“, na vysoce specializovaných pracovištích navíc i imunoelektronová mikroskopie [Seldin, 2006; Lachmann, 2002; Dispenzieri, 2012] (viz kap. 4.2.1 Diagnostika amyloidu z pohledu patologa a kap. 4.3 Speciální diagnostické techniky u systémové AL amyloidózy).
- Monoklonální charakter plazmocelulárních elementů u ALA lze prokázat detekcí intaktní molekuly MIg a/nebo zvýšené produkce VLŘ MIg s patologickým indexem κ/λ (viz kap. 4.2.2 Průkaz MIg a VLŘ v séru a/nebo moči). Klíčovým znakem systémové ALA je nejčastěji zvýšená hladina VLŘ typu λ [Gertz, 2011]. Samotný průkaz MIg ovšem nepotvrdí s jistotou, že jde o AL typ amyloidu. Přímým průkazem monoklonality plazmocytů je stanovení poměru plazmocytů s expresí LŘ κ/λ pomocí imunohistochemického vyšetření trepanobioptického vzorku kostní dřeně nebo analýzou aspirátu KD pomocí M-FC (multiparametrické průtokové cytometrie) (viz kap. 4.2.1 Diagnostika amyloidu z pohledu patologa a 4.3.3 Vyšetření klonality plazmocytů pomocí multiparametrické průtokové cytometrie).
Vzhledem ke složitosti časné diagnostiky tak proměnlivého onemocnění, jakým je systémová ALA, byla pro potřeby klinické praxe dohodnuta v rámci „10th International symposium on amyloid and amyloidosis, Tours 2004“ kritéria přesně definující klinicko-laboratorní známky orgánového postižení. Tato kritéria byla v roce 2010 částečně revidována (tab. 6).
![Průkaz postižení orgánů při systémové AL amyloidóze [Gertz, 2005; Gertz et Merlini, 2010] (Consensus opinion from the 10th International
symposium on amyloid and amyloidosis, Tours, 2004)](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/0a08056d4b2a47bc583c5cf5843810ed.png)
GIT – gastrointestinální trakt, ECHO – echokardiografie, NT-proBNP – propeptid mozkového natriuretického peptidu, AF – alkalická fosfatáza, DKK – dolní končetiny
5.2 Diagnostická kritéria postižení jednotlivých orgánových systémů u systémové AL amyloidózy
5.2.1 Diagnostika amyloidové kardiomyopatie
Laboratorní vyšetření. Stanovení kardiálních biomarkerů je nedílnou součástí diagnostického algoritmu u nemocných s ALA. Obvykle jsou vyšetřovány hladiny troponinu T (TnT) a NT-pro brain natriuretického peptidu (NT-proBNP), ojediněle i troponinu I (TnI), laboratorně standardizovanými metodami [Pika, 2008]. Hladina TnT je citlivým ukazatelem myokardiálního postižení a její hodnota stanovená v době diagnózy (> 0,035 µg/l) má úzký vztah k prognóze, proto se stala jedním z kritérií stratifikačního systému systémové ALA vypracovaného na Mayo Clinic [Kristen, 2010; Dispenzieri, 2003; Dispenzieri, 2004b]. Přetrvávání zvýšené hladiny TnT signalizuje pokračující ztrátu kardiomyocytů, zato účinná terapie je spojena s různě významným poklesem. Rovněž hladina NT-proBNP má prognostický a stratifikační potenciál (> 332 ng/l) a při jeho normální hladině je amyloidová kardiomyopatie prakticky vyloučena [Dispenzieri, 2004b; Palladini, 2003]. Vzhledem k tomu, že při účinné léčbě dochází k poklesu hladin obou srdečních biomarkerů, jsou používána jako kritéria pro hodnocení orgánové kardiologické remise [Comenzo, 2012]. Limitací stanovení NT-proBNP je přetrvávání zvýšených hladin při terapii imunomodulačními léky (IMIDy) a stejně jako v případě TnT jsou i hladiny NT-proBNP ovlivněny případnou renální nedostatečností [Dispenzieri, 2010b]. Proto se v této situaci jeví jako výhodnější stanovení samotného BNP (brain natriuretic peptide), který není renální nedostatečností ovlivněn. Vzhledem k tomu, že metoda stanovení není standardizovaná, je nutné použít pro monitoraci stavu vždy stejnou laboratoř [Palladini, 2012].
Paraklinická vyšetření. Mezi základní a neopomenutelná vyšetření u ALA patří elektrokardiografické vyšetření (EKG). V případě pokročilé amyloidové kardiomyopatie je typickým nálezem snížená voltáž v končetinových svodech < 5 mm a obraz QS ve svodech V1–3 (tzv. obraz pseudoinfarktu), poměrně častým nálezem je záchyt arytmií či AV blokád [Fikrle, 2013; Murtagh, 2005]. Stěžejním vyšetřením v diagnostice a sledování srdečního postižení při amyloidóze je echokardiografie a MR (kap. 4.2.3 Zobrazovací techniky u systémové AL amyloidózy). Endomyokardiální biopsie (EMB) je indikována v případě podezření na srdeční amyloidózu při negativitě necílené biopsie. Stejně tak lze zvážit provedení EMB při přítomnosti atypických faktorů, např. arteriální hypertenzi či šíři interventrikulárního septa > 12 mm. O závažnosti postižení vegetativního nervového systému může do jisté míry informovat test variability srdeční frekvence (Ewingův test) [Reyners, 2002].
5.2.2 Diagnostika amyloidové nefropatie
Ledviny jsou v případě systémové ALA nejčastěji postiženým orgánem (50–80 %) projevujícím se v úvodu nápadnou proteinurií, případně i vzestupem sérového kreatininu. Neselektivní proteinurie dosahuje většinou intenzity nefrotického syndromu (NS) (2,5 až 30 g/24 hod.), kdy většinu tvoří albumin. Nezbytnou součástí vyšetření je nejen standardní, ale i imunofixace (IFE) moči zaměřená na detekci MIg a VLŘ, tj. Benceovu-Jonesovu bílkovinu. Laboratorní vyšetření musí obsáhnout i ostatní atributy NS, včetně detekce hyperlipoproteinemie a dalších obvyklých komplikací. Minimální proteinurie se vyskytuje pouze v případech, kdy depozita amyloidu jsou omezena na tubuly, intersticium a cévní složku ledvinové tkáně.
Glomerulární filtrace (GF). Vyšetření GF je standardním vyšetřením, neboť u amyloidové nefropatie dochází k jejímu rychlému poklesu s častým vývinem ESRD (end-stage renal disease) [Dember, 2010]. I když rychlost poklesu GF je u ALA individuálně značně odlišná, u většiny nemocných postupně stále klesá zejména v případě prevalence glomerulárních amyloidových depozit nad depozity v tubulointersticiální oblasti. V pozdní fázi nemoci lze zaznamenat v důsledku poškození cévní stěny a zvýšené propustnosti bazální membrány glomerulu mikroskopickou hematurii. I v případě ledvin platí, že porucha orgánové funkce závisí především na toxicitě fibrilárních prekurzorů, tj. prekurzorových proteinů (amyloidogenních VLŘ) a jejich oligomerů než na rozsahu amyloidových depozit [Snanoudj, 2004; Dember, 2010]. Při hodnocení poklesu GF je nutné zohlednit i pokles prokrvení ledvin způsobený snížením srdečního výkonu, poklesem krevního tlaku a intravaskulárního objemu krve, závažností autonomní dysfunkce či předchozí aplikací kontrastní látky. K akutnímu, obvykle jen přechodnému, snížení GF s prohloubením retence dusíkatých katabolitů vyžadujícím dialyzační léčbu dochází asi u 20 % nemocných po vysokodávkované léčbě následované autologní transplantací, ale třeba i po léčbě lenalidomidem. Při hodnocení výsledku vyšetření GF je nutné počítat s okolností, že dosažení hematologické remise s potlačením produkce amyloidogenních LŘ vede obvykle pouze ke stabilizaci než ke zlepšení renální funkce. Vyšetření GF v klinické praxi má v případě systémové ALA určité úskalí, neboť hladina kreatininu v séru může v případě ztráty svalové tkáně převyšovat stupeň snížení GF. Je nutné počítat s jistou nepřesností výpočtu GF s pomocí clearence kreatininu i Cockroftovy-Gaultovy rovnice, neboť v případě NS je hodnocení objemu svalové hmoty nepřesné. V hraničních situacích lze využít vyšetření GF s pomocí cystatinu a radionuklidových metod.
Tubulární funkce, sonografie a biopsie ledviny. Vyšetření tubulárních funkcí je plně indikováno vzhledem k relativně častému výskytu funkčních tubulárních poruch, avšak úplný obraz Fanconiho syndromu (postižení proximálního tubulu) nebo nefrogenního typu diabetes insipidus (postižení distálního tubulu) je spíše výrazem současné koincidence s MM [Ryšavá, 2013]. Součástí základního diagnostického algoritmu je i sonografie ledvin, neboť u většiny nemocných se setkáváme v důsledku renální depozice amyloidu se symetrickým zvětšením a změnou echogenity [Krejčí, 2011]. V případě absence pozitivity necílené biopsie je rozhodnutím volby cílená biopsie ledviny, nejčastěji dominantně postiženého parenchymatózního orgánu, vyznačující se 90–95% záchytem amyloidu a umožňující přesné rozpoznání umístění depozit a charakteru disrupce architektury ledvinové tkáně [Ryšavá, 2013].
5.2.3 Kritéria postižení ostatních orgánů u AL amyloidózy
V rámci systémové AL amyloidózy lze vedle dominantně postižených ledvin a srdce odhalit i postižení dalších orgánů a orgánových systémů:
Nervový systém. Zde se postižení projevuje přítomností distální symetrické, senzomotorické periferní neuropatie DKK a postižením autonomního nervového systému. Rozpoznání distální smíšené periferní neuropatie DKK je založené na rozboru klinického obrazu (viz kap. 3.2.3 Periferní a autonomní neuropatie) a pečlivém neurologickém vyšetření doplněném o EMG, i když jeho nedostatkem je poměrně nízká citlivost. Individuálně lze použít histologické vyšetření surálního nervu. Rozpoznání autonomní neuropatie je založeno především na rozboru klinického obrazu (viz kap. 3.2.3 Periferní a autonomní neuropatie), případně doplněném o testy používané ke zhodnocení vegetativní dysfunkce.
Gastrointestinální trakt (GIT). Klinické projevy přímé infiltrace stěny zažívací trubice amyloidem vedoucí k poruše motility, erozivní amyloidové enteropatii či malabsorpci jsou indikací k endoskopickému vyšetření GIT s odběrem materiálu na histologické vyšetření. Podezření na postižení jater, vyznačující se vedle hepatomegalie ověřené sonograficky nebo pomocí CT (šíře > 15 cm) přítomností portální hypertenze se splenomegalií a více než 1,5násobkem zvýšené jaterní frakce alkalické fosfatázy (AF), je indikací k provedení biopsie jater.
Postižení plic. Jeho průkaz je založen na provedení konvenčního radiografického vyšetření plic, případně i vysoce citlivého HRCT (high-resolution CT) odhalujícího i jinak nerozpoznatelné postižení plic a pleury depozity amyloidu. V případě zmnožené retikulonodulární kresby (difuzní intersticiální amyloidóza), ložiskové léze (coin lesion, resp. amyloidom), pleurálního výpotku při postižení pleury může správné diagnóze napomoci transbronchiální nebo video-asistovaná torakoskopická biopsie. V odhalení depozit v bronchiálním stromu (tracheobronchiální amyloidóza) se uplatňuje bronchoskopie s odběrem materiálu na histologii a spirometrické vyšetření včetně vyšetření difuzní plicní kapacity.
Postižení pohybového aparátu. V případě postižení svalstva a kloubního systému (viz kap. 3.2.5 Postižení ostatních orgánů a tkání) lze využít vedle vyšetření svalových enzymů (kreatinkináza, myoglobin v séru) i MR a biopsii svalu, v případě amyloidové artropatie biopsii synovie či histologické vyšetření tkáně získané při operaci karpálního tunelu.
Postižení krvetvorného a koagulačního systému. Vyšetření krevního obrazu může odhalit anemii chronických chorob a zvýšení počtu krevních destiček. Koagulační vyšetření může zaznamenat zvýšení fragility kožních cév, prodlouženou krvácivost a aPTT spolu se sníženou hladinou faktoru X, ke které dochází v důsledku vazby tohoto koagulačního faktoru na amyloid [Mumford, 2000].
Postižení ostatních orgánů a tkání. Pro jeho potvrzení je vedle zhodnocení klinického obrazu (viz kap. 3.2.5 Postižení ostatních orgánů a tkání) vždy nutný odběr tkáně k histologickému vyšetření, např. excize svaloviny jazyka při makroglosii, biopsie kůže v případě kožních depozit amyloidu, histologie lymfatické uzliny při lymfadenomegalii.
5.3 Diagnostický algoritmus systémové AL amyloidózy
V klinické praxi lze přistupovat k nemocnému s podezřením na systémovou amyloidózu podle „diagnostického algoritmu“, zohledňujícího i další typy amyloidóz, než je ALA (tab. 7). Vždy je ale nutné splnit čtyři základní diagnostické kroky [Dispenzieri, 2012], tj.:
- pomyslet na možnost ALA
- potvrdit přítomnost amyloidózy s pomocí tkáňové biopsie
- určit prekurzorový protein
- posoudit rozsah orgánového postižení

6 DIFERENCIÁLNÍ DIAGNÓZA SYSTÉMOVÉ AL AMYLOIDÓZY
V případě histologického průkazu amyloidózy je nutné odlišení systémové ALA od formy ložiskové a AL amyloidózy provázející MM nebo jiný B-lymfoproliferativní stav (kap. 8 Systémová AL amyloidóza a mnohočetný myelom). Ložiskový typ AL amyloidózy se vyznačuje jistou predilekční lokalizací, nepřítomností MIg, normální hodnotou VLŘ v séru a nepřítomností monoklonální populace plazmocytů v kostní dřeni (kap. 10 Ložisková AL amyloidóza). Reaktivní AA amyloidóza navazuje na předchozí dlouhodobou přítomnost základního vyvolávajícího onemocnění (např. revmatoidní artritida), jsou zvýšeny hodnoty reaktantů akutní fáze a sérového amyloidu A (SAA), pozitivita imunohistologického vyšetření s použitím specifické monoklonální protilátky. Vzácný hereditární ATTR typ amyloidózy lze rozpoznat s pomocí vysoce specializovaných metod (DNA analýza, hmotnostní spektrometrie, imunoelektronová mikroskopie) a je nutné mít na paměti i možnou přítomnost MIg (v 5–10 %) [Bird, 2004]. U nemocných v dlouhodobé dialyzační léčbě se ojediněle vyskytuje hemodialyzační (Aβ2M) amyloidóza. U starších mužů s dominantním postižením srdce a s projevy městnavé srdeční slabosti a/nebo poruchami rytmu je nutné pátrat po amyloidóze senilní. Musíme mít na paměti, že na možnost systémové ALA je nutné pomýšlet u všech jedinců s MGUS, a před stanovením definitivní diagnózy provést pečlivé vyšetření podle diagnostického algoritmu ALA, případně i opakovaně. Je nutné odlišit rovněž monoclonal gammopathy of renal significance (MGRS) provázející ojediněle MGUS s přítomností LŘ s vysokým nefrotoxickým potenciálem [Leung, 2012], ale i onemocnění s depozity lehkých řetězců (LCDD – light chain deposition disease) [Ščudla, 2012] nebo z depozice těžkých řetězců imunoglobulinu (HCDD – heavy chain deposition disease), případně i periferní neuropatii s přítomností MIg [Bird, 2004]. Stejně jako u ALA jsou v případě LCDD postiženy nejčastěji ledviny (renální insuficience), méně časté je zvětšení jater s poruchou jaterní funkce a/nebo postižení srdce s projevy městnavé srdeční slabosti. Precizní zařazení zjištěné amyloidózy je nezbytné nejen z hlediska odlišné klinické manifestace, průběhu a prognózy jednotlivých typů, ale především z hlediska poskytnutí účelné, pro jednotlivé typy amyloidóz specifické, terapie [Falk, 1997; Ščudla, 2009].
7 PROGNOSTICKÉ FAKTORY A KLINICKÁ STRATIFIKACE SYSTÉMOVÉ AL AMYLOIDÓZY
7.1 Prognostické faktory
Prognóza nemocných se systémovou ALA je variabilní, stále ale nepříznivá, a to zejména pokud nebyla včas zahájena adekvátní terapie [Kyle, 1997]. V historických studiích byl medián celkového přežití 15–18 měsíců, ale v případě nemocných se závažným postižením srdce manifestujícím se městnavou srdeční slabostí a/nebo synkopálními stavy pouze 4–6 měsíců [Kyle 1995a; Dispenzieri, 2010a]. Nemocní s projevy závažné amyloidové kardiomyopatie se vyznačují vysokým rizikem náhlé smrti [Gertz, 1999, 2011]. V současnosti, díky časnější diagnóze a nesporným pokrokům v léčbě, se délka celkového přežití nemocných s ALA podstatně zlepšila a v posledním desetiletí dosáhla v 4letém odstupu od diagnózy dvojnásobku [Kumar, 2010]. Prognóza nemocných se systémovou ALA závisí zejména na dosažení rychlé suprese syntézy monoklonálních LŘ a na stabilizaci, případně zlepšení, srdeční funkce [Wechalekar, 2009].
Excelentními prediktory prognózy ALA jsou sérové hladiny solubilních srdečních biomarkerů (troponin T, troponin I, NT-proBNP a BNP) [Palladini, 2003; Dispenzieri, 2003, 2004b], které se staly východiskem prognostického stratifikačního systému systémové ALA (viz kap. 7.2 Klinická stratifikace). Hladiny NT-proBNP > 8500 ng/l spolu s nálezem hodnot systolického tlaku < 100 mm Hg identifikují nemocné s vysokým rizikem časného úmrtí [Wechalekar, 2013].
Významnými prognostickými ukazateli ALA jsou i charakteristiky plazmocelulárního klonu, jmenovitě stupeň plazmocytózy KD (> 10 %) či procento aberantních plazmocytů při použití multiparametrické průtokové cytometrie [Gertz, 1989; Dispenzieri, 2010a; Paiva, 2011]. Nemocní s přítomností t(11; 14) mají horší prognózu, pokud jsou léčeni režimy na bázi bortezomibu, naopak profitují z léčby vysokodávkovaným melfalanem [Bochtler, 2015, 2016]. Nemocní se ziskem v oblasti 1q21 neprofitují z léčby kombinací melfalanu a dexamethasonu [Bochtler, 2014].
Vysoká hodnota dominantního VLŘ v séru a patologie κ/λ indexu vyšetřené v době stanovení diagnózy mají úzký vztah k prognóze [Dispenzieri, 2006]. Prognosticky příznivou skupinu tvoří nemocní s nízkou náloží VLŘ (dFLC < 50 mg/l) v době diagnózy. U těchto nemocných bývá menší procento a závažnost kardiálního postižení, naopak bývá častější renální postižení s vyšším stupněm proteinurie. Nemocní se vyznačují delším celkovým přežitím [Dittrich, 2017; Milani, 2017].
Z ukazatelů vyšetřených po ukončení terapie je nejvýznamnější hloubka orgánové léčebné odezvy a hloubka hematologické odezvy, zejména dosažení kompletní remise (CR) založené na vymizení M-proteinu v séru a/nebo v moči vyšetřeného IFE, normalizaci VLŘ v séru včetně indexu κ/λ a vymizení plazmocelulárního klonu v KD [Gertz, 2007].
7.2 Klinická stratifikace
V roce 2004 byl navržen stážovací systém rozdělující nemocné se systémovou AL amyloidózou na podkladě hodnot srdečních biomarkerů do tří stadií [Dispenzieri, 2004b], neboť závažnost postižení srdce je i u asymptomatických jedinců stěžejním prognostickým faktorem [Palladini, 2010b; Comenzo, 2012] (tab. 8).
![„Mayo Clinic“ stážovací systém systémové AL amyloidózy
[Dispenzieri, 2004b]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/ff03435a04192280062234ad7f6f1352.png)
V roce 2012 byl na Mayo Clinic vypracován tzv. „revidovaný prognostický stážovací systém“ založený na hladinách cTnT, NT-proBNP a VLŘ v séru (tab. 9). To vedlo ve srovnání s původním stratifikačním systémem ke zlepšení hodnocení dlouhodobého prognostického výhledu u nemocných léčených standardní chemoterapií i ASCT [Gertz, 2011; Kumar, 2012].
![Revidovaný „Mayo Clinic“ stážovací systém systémové AL amyloidózy [Kumar, 2012]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/b14ba436dad5cc490c2c8fc677f8e2f9.png)
cTnT – c-troponin, NT-proBNP – N-terminální pro-brain natriuretický peptid (N-terminal pro-brain natriuretic peptide), FLC – volné lehké řetězce imunoglobulinu (free light chain), OS – celkové
přežití (overall survival)
Míra poškození srdce je prognosticky nejzávažnějším faktorem, který je nutné zohlednit i při stanovení intenzity léčby. U nemocných s vysokým kardiálním rizikem (troponin T > 0,06 ng/ml nebo NT-proBNP > 5000 pg/ml u nemocných s normální funkcí ledvin) není doporučeno provedení autologní transplantace z důvodu vysoké časné peritransplantační mortality [Gertz, 2016]. Proto byl navržen a validován Evropský stážovací systém pokročilého srdečního postižení [Wechalekar, 2013] (tab. 10).
![Evropský stážovací systém pokročilého srdečního postižení při
systémové AL amyloidóze [Wechalekar, 2013]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/08ae7e675a623a5059b6924c9513c809.png)
8 SYSTÉMOVÁ AL AMYLOIDÓZA A MNOHOČETNÝ MYELOM
Dle míry infiltrace kostní dřeně klonálními plazmocyty rozlišujeme nemocné s AL amyloidózou (< 10 % plazmocytů) a nemocné s mnohočetným myelomem a asociovanou AL amyloidózou (≥ 10 % plazmocytů). Někteří jedinci, u nichž se v průběhu sledování rozvinou příznaky systémové ALA, jsou dlouhodobě vedeni jako MGUS. U malé části nemocných s MGUS (~ 1 až1,5 %/rok) dochází k vývinu systémové ALA, případně MM nebo WM, neboť jde ve všech případech o klonální proliferaci elementů plazmocelulární, případně lymfoplazmocytární linie [Kyle, 2010]. Současný výskyt systémové ALA a MM se pozoruje u 10–15 % nemocných [Fielder, 1986; Kyle, 1995b], ovšem systémová ALA je asi 10krát méně častá nežli MM. Asi u 1 % nemocných s nově diagnostikovaným symptomatickým i asymptomatickým MM lze cíleným vyšetřením rozpoznat asymptomatickou formu systémové ALA [Siragusa, 2011]. LŘ produkované maligními monoklonálními plazmocyty nabývají v důsledku polymerizace a částečné degradace v elementech monocytárně-makrofágového systému amyloidogenní potenciál. V klinické praxi diagnóza MM obvykle předchází nebo odpovídá době rozpoznání ALA. Z hlediska strategie léčby a hodnocení prognózy je vždy nutné v této situaci rozhodnout, který stav je „dominantní“. Naopak výjimečně, v 0,4 %, dochází k manifestaci MM ve více než šestiměsíčním odstupu (odložená progrese) od diagnózy ALA, a to obvykle u relativně indolentních forem nemoci bez postižení srdce, ledvin a jater, vyznačujících se dlouhým průběhem [Rajkumar, 1998]. V této situaci je rozpoznání symptomatického MM založeno na přítomnosti anemie, > 30 % plazmocytů v KD, hyperkalcemii a bolestech skeletu s přítomností osteolytických lézí, ale i na odhalení dalších nálezů včetně cytogenetických změn, pozorovaných obvykle při maligní transformaci MGUS v MM [Alpers, 1986]. Na vývin ALA je nutné vždy pomýšlet především v případě výskytu projevů, které obvykle nepatří do klinického obrazu MM, např. závažné postižení srdce s projevy restriktivní kardiomyopatie, syndrom karpálního tunelu, makroglosie, nefrotický syndrom, projevy zejména autonomní neuropatie, nález kožních ložisek amyloidu aj. (kap. 3 Klinický obraz systémové AL amyloidózy). V případě přítomnosti plně vyjádřených projevů amyloidové kardiomyopatie je prognóza nemocných se současným MM značně nepříznivá, s celkovým přežitím ~ 9 měsíců [Madan, 2010]. Při nepřítomnosti závažného postižení srdce nebo jiného parenchymatózního orgánu je možná dobrá odezva k chemoimunoterapii s dosažením délky přežití obdobné, jako je tomu u samotného MM [Fielder, 1986].
9 DOPORUČENÍ PRO DIAGNOSTIKU AL AMYLOIDÓZY
- Systémová AL amyloidóza je vzácné hematologické onemocnění patřící mezi monoklonální gamapatie
- Optimální péče o nemocné s ALA je založena na časné diagnóze s vyhodnocením závažnosti orgánového postižení, účinné terapii zaměřené zejména na snížení produkce amyloidogenních lehkých řetězců a léčbě podpůrné.
- V 12–20 % je asociována s jinou formou monoklonální gamapatie či B-lymfoproliferativního onemocnění.
- Amyloidóza musí být vždy histologicky verifikována spolu s potvrzením AL typu amyloidu.
- Podkladem onemocnění je depozice amyloidu v orgánech a tkáních, což vede k jejich funkčnímu postižení, ev. selhání. Pečlivé posouzení orgánového postižení je základním pilířem diagnostiky a stratifikce nemocných.
- Detekce a kvantifikace monoklonálního imunoglobulinu a zejména hladin volných lehkých řetězců patří mezi stěžejní aspekty diagnostiky a sledování nemocných se systémovou ALA.
10 LOŽISKOVÁ AL AMYLOIDÓZA
10.1 Etiopatogeneze, diagnostika a klinický obraz
Ložisková ALA se vyznačuje pouze lokální depozicí amyloidu bez známek systémového výskytu. Amyloid je deponován pouze v místě syntézy amyloidogenního proteinu, tj. v oblasti ložiskového nahromadění monoklonálních plazmocytů, přičemž prekurzorové amyloidogenní LŘ nejsou transportovány do krevního řečiště. Existují však i příklady terčové depozice cirkulujícího MIg u systémové ALA ve specifickém orgánu v důsledku „tkáňového tropismu“ [Benson, 1995]. Incidence ložiskové AL amyloidózy odpovídá 10–20 % všech ALA a vyskytuje se především v 6. a 7. životní dekádě [Kyle, 1975]. I když byla odkryta již řada patofyziologických mechanismů, její etiologie není doposud vyřešena. Amyloidogenní LŘ jsou vytvářeny populací klonálních plazmocelulárních nebo lymfoplazmocytárních elementů obklopujících formou nodulárních nebo difuzních infiltrátů ložiska amyloidu [Hamidi, 1999]. U většiny případů nelze zachytit MIg v séru a/nebo v moči, stejně tak ani zvýšení hladin VLŘ. Ložisková amyloidóza se v naprosté většině případů vyznačuje imunohistochemickou příslušností k AL typu. AA i transthyretinový typ amyloidu se objevují v ložiskové formě velmi vzácně, např. výjimečné ložiskové postižení hrtanu v případě hereditární systémové amyloidózy v rámci varianty apolipoproteinu AI. Lokalizovaná ALA se vyskytuje nejčastěji v oblasti horních dýchacích cest, zejména v nazofaryngu, v urogenitální oblasti a v kůži, je ale známo i postižení orbity, GIT, páteře, prsů a dalších měkkých tkání, zcela vzácně ve formě kostních lézí v oblasti páteře. Při objevení amyloidového ložiska je nutné nejprve vyloučit příslušnost k systémové formě ALA. Klinický průběh je většinou benigní a projevuje se lokálními příznaky, ojediněle však může vyústit v závažné narušení funkce postiženého orgánu. Postižení laryngu včetně hlasových vazů se projevuje vedle lokálního krvácení především dysfonií, ojediněle i obstrukcí s projevy inspirační dechové tísně včetně stridoru. K objasnění rozsahu procesu je nezbytná laryngoskopie s odběrem materiálu na histologické vyšetření, případně CT vyšetření [Ma, 2005]. Postižení hrtanu se projevuje přítomností difuzních subepiteliálních depozit (difuzní typ) nebo diskrétních či polypoidních nodulárních depozit (nodulární typ). Ložisková amyloidóza spojivek bývá zaměňována za maligní lymfom. V případě postižení uropoetického systému jde téměř výhradně o projev ložiskové, nikoliv systémové, ALA. Může být postižena ledvinová pánvička, močovod, močový měchýř, uretra a jde prakticky bezvýhradně o projev ložiskové, nikoliv systémové, AL amyloidózy. V klinickém obraze může dominovat vedle známek dysurie a recidivujících zánětů makroskopická hematurie. Diagnóza ložiskové ALA je založena na urologickém vyšetření včetně biopsie a CT-urografie. Kožní a podkožní depozita amyloidu se projevují ve formě makulárních pigmentových eflorescencí, papulárních a nodulárních útvarů v oblasti zad, hrudníku, šíje a rtů, a mohou perzistovat řadu let bez jakékoliv změny. Ložisková depozita amyloidu se mohou rovněž vyskytovat i v tkáni extramedulárního plazmocytomu, karcinomu nasopharyngu, prsu, močového měchýře a medulárního karcinomu štítné žlázy [Munichor, 2000; Chiang, 2013; Gupta, 2012].
Klinický obraz ložiskové AL amyloidózy, její průběh, prognóza a léčba se zcela zásadně liší od formy systémové, proto je před stanovením definitivní diagnózy nutné pečlivé rozlišení obou stavů. Přechod ložiskové ALA ve formu systémovou je velmi vzácný a byl popsán jen raritně [Paccalin, 2005].
10.2 Léčba ložiskové AL amyloidózy
V případě místních projevů ložiskové ALA se uplatňuje opakované chirurgické odstranění a mikroabraze místních ložiskových mas v rámci laryngoskopie a bronchoskopie s eventuální dilatací a případným zavedením silikonového stentu. Velmi příznivé zkušenosti jsou zejména v laryngeální oblasti s použitím Nd:YAG (neodymium-doped yttrium aluminum garnet) nebo CO2 laseru vedoucí k dlouhodobé stabilizaci procesu [Motta, 2003; Kurrus, 1998]. V případě tracheobronchiální amyloidózy způsobující závažné zúžení dýchacích cest s výraznou inspirační dechovou tísní je nutná tracheotomie. Vzhledem k radiosenzitivitě plazmocelulárních elementů a tím i možnosti lokálního potlačení produkce amyloidogenních LŘ lze individuálně úspěšně využít zevní ložiskovou aktinoterapii formou 10 až 12 frakcí v celkové dávce 20–45 Gy. Té zpravidla předchází lokální odstranění amyloidových mas [Kalra, 2001; Kurrus, 1998; Neuner, 2012]. V případě postižení močových cest lze vedle transuretrální resekce vyzkoušet i rozpuštění fibril amyloidu opakovanou instilací dimetylsulfoxidu do močového měchýře a u malých lézí ošetření laserem. U kožních forem se vedle chirurgické excize a ošetření laserem může uplatnit intralezionální aplikace kortikosteroidů a dermabraze. Systémová chemoterapie včetně léčby kortikosteroidy se pro svou neúčinnost u ložiskové ALA nedoporučuje [Bird, 2004; Dominguez, 1996]. Ložisková ALA je v podstatě nevyléčitelná, a většinou proto vyžaduje opakovanou lokální léčbu. Přestože má ve srovnání se systémovou ALA mnohem příznivější prognózu, je i ložisková ALA provázena významnou morbiditou, která závisí na závažnosti postižení a orgánové lokalizaci [Buadi, 2010].
Použitá literatura
1. Benson MD. Amyloidosis. In: Scriver CR, et al. The metabolic and molecular bases of inherited disease. 7th ed. New York: McGraw Hill 1995; 4159–4191.
2. Bird J. Guidelines on the diagnosis and management of AL amyloidosis. Br J Haematol 2004; 125 : 107–137.
3. Buadi F. Localized amyloidosis. In: Gertz MA, Rajkumar SV. Amyloidosis: diagnosis and treatment. 1st ed. New York: Springer Science 2010; 95–106.
4. Dominguez W, Weinberg P, Claros P, et al. Primary localized nasopharyngeal amyloidosis. A case report. Int J Pediatr Otorhinolaryngol 1996; 36 : 61–67.
5. Gupta P, Hanamshetti S, Kulkarni JN. Primary amyloidosis with high grade transitional cell carcinoma of bladder. J Cancer Res Ther 2012; 8(2): 297–299.
6. Hamidi AK. Organ-specific (localized) synthesis og Ig light chain amyloid. J Immunol 1999; 162 : 556–560.
7. Chiang D, Lee M, Germaine P, et al. Amyloidosis of the Breast with Multicentric DCIS and Pleomorphic Invasive Lobular Carcinoma in a Patient with Underlying Extranodal Castleman’s Disease. Case Rep Radiol 2013; 2013 : 190856.
8. Kalra S. External-beam radiation therapy in the treatment of difuse tracheobronchial amyloidosis. Mayo Clin Proc 2001; 76 : 853–856.
9. Kurrus JA, Hayes JK, Hoidal JR. Radiation therapy for tracheobronchial amyloidosis. Chest 1998; 114 : 1489–1492.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine (Baltimore) 1975; 54 : 271–299.
11. Ma L. Primary localized laryngeal amyloidosis: report of 3 cases with long-term follow-up and review of the literature. Arch Pathol Lab Med 2005; 129 : 215–218.
12. Motta G, et al. CO (2)-laser treatment of laryngeal amyloidosis. J Laryngol Otol 2003; 117 : 647–650.
13. Munichor M, et al. Localized amyloidosis in nasopharyngeal carcinoma diagnosed by fine needle aspiration and electron microscopy. A case report. Acta Cytol 2000; 44 : 637–638.
14. Neuner GA, Badros AA, Meyer TK, et al. Complete resolution of laryngeal amyloidosis with radiation treatment. Head Neck 2012; 34 : 748–752.
15. Paccalin M. Localized amyloidosis: a survey of 35 French cases. Amyloid 2005; 12 : 239–245.
11 CÍLE LÉČBY A HODNOCENÍ LÉČEBNÉ ODPOVĚDI U AL AMYLOIDÓZY
Cílem léčby AL amyloidózy je eliminace produkce amyloidogenních lehkých řetězců klonálními plazmocyty. Redukce, resp. eliminace, nálože amyloidogenních lehkých řetězců a současně produkující klonální plazmocelulární populace je podmínkou dosažení hematologické remise onemocnění. Dosažení hematologické remise onemocnění je předpokladem pro dosažení orgánových léčebných odpovědí při postižení jednotlivých orgánových systémů, tedy zlepšení jejich funkce a prodloužení přežití nemocných [Merlini, 2006]. Palladini et al. definoval velmi dobrou parciální remisi (VGPR) onemocnění jako minimální míru hematologické léčebné odpovědi, která by měla být léčbou dosažena (úroveň důkazu IIa, stupeň doporučení B) [Palladini, 2012a].
Při hodnocení léčby AL amyloidózy se udává míra hematologické léčebné odpovědi, která je definována na základně stanovení hladin MIg v séru či moči a/nebo VLŘ a na základě vyšetření kostní dřeně [IMWG, 2003; Kyle, 2009]. Dále se stanovuje tzv. orgánová léčebná odpověď, jež je definována dalšími laboratorními a paraklinickými údaji vyjadřujícími funkční schopnost orgánů postižených depozity amyloidu [Gertz, 2005, 2010b; Comenzo, 2012]. Definici hematologických léčebných odpovědí uvádí tabulka 11, definice orgánových odpovědí jsou uvedeny v tabulce 12.
![Definice hematologických léčebných odpovědí u pacientů s AL amyloidózou [Gertz, 2005, 2010b; Comenzo, 2012]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/c754edee3857d4ccd2619864c12f7728.png)
![Definice orgánových léčebných odpovědí u pacientů s AL amyloidózou [Gertz, 2005, 2010b; Comenzo, 2012]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/5e098661127557564175f06b16681276.png)
12 LÉČEBNÉ STRATEGIE U AL AMYLOIDÓZY V ROCE 2019
AL amyloidóza je vzácné hematologické onemocnění a řada doporučení pro léčbu je vytvořena zpravidla na základě nekontrolovaných léčebných studií. U nemocných, u kterých je AL amyloidóza asociována s mnohočetným myelomem, se doporučovaná léčebná schémata neliší od terapeutických doporučení pro toto onemocnění s respektováním indikačních kritérií, kontraindikací a cíle dosažení alespoň velmi dobré parciální remise [Hájek, 2018]. V současné době není zcela jasný mezinárodní konsenzus stran optimální terapie pro nově diagnostikované pacienty s AL amyloidózou. Léčebná strategie je zaměřena na eliminaci fibrilárních prekurzorů, resp. volných lehkých řetězců, které jsou schopny infiltrovat extracelulární prostor cílových orgánů a po jistém čase způsobit jejich závažnou dysfunkci. Ta je především v případě poškození srdce velmi často fatální [Ryšavá, 2018]. AL amyloidóza je zpravidla velmi senzitivní onemocnění, léčba nemusí být zpravidla tak intenzivní jako například u mnohočetného myelomu. Tento názor podporuje i skutečnost, že nemocní s AL amyloidózou jsou častěji náchylní k závažným komplikacím, což souvisí s poškozením orgánů často vstupně skrytým. Mezi experty se stále více prosazuje názor, že by v případě nové diagnózy (ale i relapsů) měla být cílem hematologická kompletní remise, tedy eradikace klonu plazmocytů s produkcí volných lehkých řetězců [Gertz, 2007]. Lze očekávat, že se zvyšujícím se počtem dostupných účinných léků bude tato léčebná strategie stále více preferovanou, třebaže stávajícím doporučením je dosažení nejméně velmi dobré parciální remise [Palladini, 2012a]. Nové léčebné možnosti umožňují nasazení jiné léčby v případě neúspěchu léčby první, což zcela jistě zvýší počet nemocných dosahujících kompletní hematologickou remisi. Je vhodné zdůraznit, že k průkazu léčebné odpovědi v případě orgánového postižení potřebujeme trpělivost, neboť někdy ji můžeme správně vyhodnotit až s odstupem 12–18 měsíců [Gavriatopoulou, 2018].
Existuje jen málo randomizovaných studií srovnávajících různé terapeutické přístupy a většina z nich nezahrnuje nejnovější léky nebo rizikovou stratifikaci. Nicméně v zahraničí je v řadě zemí péče o nemocné s AL amyloidózou směřována do velkých center a lze z části navázat na doporučení těchto pracovišť. Tato doporučení jsou běžně odbornou veřejností celosvětově akceptována a je nutné podotknout, že doporučení významných světových center se obsahově výrazně neliší [Wechalekar, 2015; Dispenzieri, 2015; Palladini, 2016]. V současnosti se v případě AL amyloidózy preferuje tzv. risk-adapted přístup, tedy léčba pacientovi „šitá na míru“ dle stratifikačních kritérií uvedených v předchozích kapitolách. Efektivní využití srdečních biomarkerů a dalších parametrů usnadnilo selekci nemocných k vysokodávkované terapii a snížilo peritransplantační mortalitu, stejně tak napomohlo i ve výběru konvenční terapie. S ohledem na relativní vzácnost onemocnění by mělo být u všech nemocných zváženo zařazení do klinických studií.
Všeobecně se u nemocných do 70 let věku a schopných podstoupit intenzivní přístup preferuje léčba vysokodávkovaným alkeranem s podporou autologních kmenových buněk, tedy autologní transplantace (ASCT). S ohledem na míru nálože klonálních buněk a hladin lehkých řetězců je na individuálním zvážení podání 2 až 4 cyklů indukční terapie s následnou stimulací a sběrem periferních kmenových buněk [Dispenzieri, 2015; Palladini, 2016]. Nicméně pouze 14–24 % pacientů je schopno tento přístup podstoupit. U nemocných s počtem klonálních buněk v kostní dřeni nad 10 %, tedy v případě asociace s mnohočetným myelomem, je indukční léčba doporučována vždy. U nemocných, kteří nejsou schopni ASCT podstoupit, je v současnosti doporučována konvenční terapie kombinací kortikoidů, alkylačních cytostatik a eventuálně některých z biologicky působících léků, nejčastěji bortezomibu. Výsledky publikovaných prací hovoří přibližně o 70% přežití v 5 letech u nemocných, kteří konvenční terapií dosáhnou kompletní remise; v případě ASCT a dosažené CR je udáváno 60% přežití v 10 letech, avšak jedná se vesměs o selektovanou skupinu nemocných [Sanchorawala, 2007a; Palladini, 2007; Gertz, 2007].
Níže uvádíme dvě léčebná doporučení z Mayo Clinic (USA) a Pavie (Itálie), která využívají risk-adapted přístup a jsou plně adaptabilní v podmínkách České republiky.
![Schéma vhodné terapie pro pacienty s nově dg. AL amyloidózou
(upraveno podle [Mayo clinic; Dispenzieri, 2015])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/dc0d5d70493fb597f15a7714865416a5.png)
![Perspektivní schéma vhodné terapie pro pacienty s nově dg.
AL amyloidózou (upraveno podle [Palladini, 2016])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/88159c532c77b1b150433cc7eb9af9c3.png)
13 AUTOLOGNÍ TRANSPLANTACE KRVETVORNÝCH BUNĚK (ASCT)
Autologní transplantace krvetvorných buněk (ASCT) byla poprvé popsána jako efektivní terapie u AL amyloidózy v roce 1998 [Comenzo, 1998]. Při použití přípravného režimu melfalan 200 mg/m2 s následnou ASCT bylo dosaženo 76 % hematologických odpovědí, 33 % CR, peritransplantační mortalita byla 12–13 % [Gertz, 2004; Skinner, 2004]. Medián přežití transplantovaných pacientů byl 4,7 roku [Sanchorawala, 2007a].
Bylo ovšem také publikováno, že pacienti s pokročilým orgánovým postižením při AL amyloidóze mají vysoké riziko úmrtí v souvislosti s transplantací (TRM), až 40 % [Moreau, 1999]. Proto bylo vyvinuto úsilí pacienty stratifikovat podle rizika před případným provedením ASCT [Gatt, 2013]. V současnosti jsou definována indikační kritéria umožňující posoudit klinický stav nemocného a určit, zda je vhodným kandidátem ASCT [Dispenzieri, 2015; Palladini, 2016]. Při výběru nemocných dle těchto indikačních kritérií se míra TRM pohybuje v centrech na úrovni 3 %. Míra léčebných odpovědí přesahuje 70 %, s 35–37 % kompletních remisí [Sanchorawala, 2015; D’Souza, 2015, Cibeira, 2011, Ryšavá, 2011, Sidiqi, 2018]. Lze konstatovat, že pro provedení ASCT je vhodná selektovaná skupina pacientů s AL amyloidózou, tito nemocní s nízkou mírou orgánového postižení však z této terapie profitují (úroveň důkazu IIa, stupeň doporučení B) [Gatt, 2013].
![Indikační kritéria pro stratifikaci nemocných k vysokodávkované
chemoterapii melfalanem s podporou autologního štěpu (úroveň
důkazu IV, stupeň doporučení C) [Dispenzieri, 2015; Palladini, 2016].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/f7ef59eede37d5d57fc20de69e51bc75.png)
13.1 Léčebná účinnost vysokodávkované chemoterapie s následnou ASCT
Pouze jediná prospektivní multicentrická randomizovaná studie, srovnávající klasickou chemoterapii melfalan a dexamethason s ASCT, byla studie francouzských autorů [Jaccard, 2007]. Do každé skupiny bylo zařazeno 50 pacientů. Medián přežití ve skupině léčené vysokodávkovanou chemoterapií byl 22,2 měsíce, medián přežití ve skupině léčené klasickou chemoterapií byl 56 měsíců. Z pacientů randomizovaných do skupiny vysokodávkované chemoterapie zemřelo ještě před podáním vysokodávkované chemoterapie celkem 10 osob, ASCT nakonec podstoupilo pouze 37 nemocných. Z nich dalších 9 zemřelo do dne 100 po transplantaci. Tato studie nepotvrdila lepší výsledky vysokodávkované chemoterapie oproti standardní chemoterapii melfalan a dexamethason. Řadou autorů je poukazováno na ne zcela optimální výběr pacientů pro ASCT v této klinické studii a dále vysoký počet zařazujících center s malým počtem nemocných (a tedy s omezenou zkušeností s metodou), což vedlo ke značné mortalitě před i po provedení ASCT [Adam, 2013].
Nicméně řada prací referuje o účinnosti ASCT, udává 32–68 % hematologických odpovědí (16–50 % CR) a 31–64 % orgánových odpovědí. Recentní práce Sanchorawala et al. a D’Souza et al. hovoří o 35–37 % kompletních remisí při 3–3,4% TRM [Sanchorawala, 2015; D’Souza, 2015].
Redukce dávky melfalanu na 140 mg/m2 je doporučována u pacientů s clearancí kreatininu < 30 ml/min [Dispenzieri, 2015]. Redukce dávky melfalanu u rizikových nemocných však snížila počet hematologických odpovědí při stejné míře TRM [Gertz, 2004], a proto není běžně doporučována (úroveň důkazu IV, stupeň doporučení C). Efektivitu a mortalitu ASCT u AL amyloidózy shrnuje tabulka 16.

** Jde o analýzu pouze těch pacientů, kteří dokončili léčbu.
CR – kompletní remise, PR – parciální remise, TRM – mortalita vázaná na léčbu
13.2 Konsolidační terapie po proběhlé autologní transplantaci
V současné době je doporučováno u nemocných, kteří podstoupili ASCT a nedosáhli alespoň velmi dobré parciální remise, zvážit podání konsolidační terapie s cílem prohloubení léčebné odpovědi (úroveň důkazu IIa, stupeň doporučení B). Doposud byla publikována jedna práce uvádějící výsledky tandemové ASCT u nemocných s AL amyloidózou, kteří po 1. ASCT nedosáhli kompletní remise. Podání 2. ASCT bylo u těchto nemocných spojeno s dosažením kompletní remise v 60 % a v 80 % přežití ve 4 letech [Sanchorawala, 2007c]. Další dvě práce referovaly o užití konsolidace pomocí kombinace thalidomidu ± dexamethasonu a bortezomibu ± dexamethasonu u nemocných, kteří nedosáhli CR [Landau, 2013; Cohen, 2016]. V obou případech bylo dosaženo prohloubení léčebné odpovědi. Landau et al. uvádí, že léčebná odpověď u pacientů, kteří nedosáhli CR po ASCT a byli léčeni kombinací bortezomibu a dexamethasonu, byla v 74 % případů kompletní [Landau, 2013].
14 KONVENČNÍ LÉČBA ZALOŽENÁ NA KOMBINACI VYSOKÝCH DÁVEK DEXAMETHASONU A ALKYLAČNÍCH CYTOSTATIK
14.1 Kombinace alkylačního cytostatika a kortikoidu
Alkylační látky jsou hlavními cytostatiky, která byla po desítky let používána u mnohočetného myelomu (MM) a byla následně terapeuticky využita i u AL amyloidózy [Kyle, 1997]. Ačkoliv perorální terapie melfalanem a prednisonem je u AL amyloidózy dobře tolerovaná a prodlužuje přežití ve srovnání s neléčenými pacienty, léčebné odpovědi jsou nízké (30 %) a jsou dosahovány pomalu, obvykle více než po roce terapie [Skinner, 1996; Kyle, 1997].
Naopak oproti prednisonu byla pozorována u AL amyloidózy efektivita dexamethasonu ve vyšší dávce [Dhodapkar, 1997]. Klinické studie kombinující orální melfalan a pulzní dexamethason (MDex) referují o 26–72 % hematologických odpovědí s přibližně 8–33 % kompletních remisí. Míra orgánových léčebných odpovědí dosahuje až 48 %. V italské studii u pacientů s AL amyloidózou, kteří nebyli vhodní pro provedení ASCT, bylo dosaženo vysoké celkové léčebné odpovědi 67 %, z toho u 33 % pacientů kompletní remise, a také byla zaznamenána nízká úmrtnost v souvislosti s terapií (4 %) [Palladini, 2004, 2007]. Medián doby do progrese pacientů léčených kombinací MDex byl 3,8 roku a medián celkového přežití 5,1 roku. Terapie MDex se stala široce akceptovanou léčbou první linie pro pacienty s AL amyloidózou, kteří nejsou vhodnými kandidáty pro provedení ASCT (úroveň důkazu IIa, stupeň doporučení B).
14.2 Nežádoucí účinky léčby a selekce nemocných
Terapeuticky problematičtí jsou pacienti s AL amyloidózou a pokročilým kardiálním postižením a obvykle špatně tolerující vyšší dávky dexamethasonu. Ovšem snížení dávky ze 40 mg na 20 mg v dny 1. až 4. cyklu bylo spojeno se signifikantně nižším počtem CR (16 % vs. 31 %) [Palladini, 2010]. Podání MDex u pacientů s pokročilým kardiálním postižením nevykazuje dobré výsledky, hematologická léčebná odpověď je udávána pouze 44 % a 26 % pacientů umírá v průběhu léčby [Dietrich, 2010]. Nicméně se zdá, že z léčebné kombinace profitují zejména nemocní ve středním riziku (stadia do IIIa). Palladini et al. uvádí 76 % hematologických odpovědí (31 %) s mediánem přežití 7,3 let pro tuto skupinu nemocných, což je srovnatelné s ASCT. Lze tedy konstatovat, že terapie MDex je bezpečná a efektivní především u pacientů se středním rizikem kardiálního postižení nebo bez kardiálního postižení, avšak není schopna překonat špatnou prognózu nemocných s pokročilým kardiálním postižením [Gatt, 2013; Palladini, 2004].
15 IMUNOMODULAČNÍ LÉKY V TERAPII AL AMYLOIDÓZY
15.1 Thalidomid v léčbě AL amyloidózy
Užití thalidomidu jako monoterapie či v kombinaci s kortikoidy a alkeranem bylo předmětem řady studií. Monoterapie thalidomidem se však neprojevila jako účinná, většinou nebylo dosaženo hematologické odpovědi (0–25 %), v kombinaci s kortikoidy pak s dosažením 48% hematologické odpovědi. Nicméně terapie byla spojena s nezanedbatelnou nehematologickou toxicitou thalidomidu (až v 65 %), zejména časté byly projevy symptomatické bradykardie a symptomy spojené s neurotoxicitou [Palladini, 2005; Seldin, 2003; Dispenzieri, 2003]. Palladini et al. užil léčebnou kombinaci thalidomidu, melfalanu a dexamethasonu ve skupině 22 nemocných s kardiálním postižením [Palladini, 2009]. Míra léčebné hematologické odpovědi dosáhla 36 %, nicméně toxicita nebyla zanedbatelná a pouze 20 % nemocných se dožilo 1 roku. Naopak britská studie referující o kombinaci thalidomidu, cyklofosfamidu a dexamethasonu u nově diagnostikovaných pacientů vypovídá o efektivitě režimu s 74% hematologickou odpovědí (21 % CR) a 77% přežitím ve 2 letech [Wechalekar, 2007]. Nicméně i tato kombinace byla spojena s vyšší mírou zejména nehematologické toxicity. Kombinaci thalidomidu, dexamethasonu a cyklofosfamidu lze ve vybraných případech zvážit jako iniciální léčebný režim, avšak s vyšším rizikem nežádoucích účinků léčby (úroveň důkazu IIa, stupeň doporučení B). V současné době jsou však režimy s thalidomidem považovány za obsolentní.
15.2 Lenalidomid v léčbě AL amyloidózy
Většina prací referujících o účinnosti lenalidomidu u nemocných s AL amyloidózou bohužel zahrnuje předléčené i nově diagnostikované pacienty, proto i interpretace dat je poměrně složitá. Kombinace lenalidomidu a dexamethasonu bývá spojena s 38–47% hematologickou odpovědí (5–16 % CR) a mediánem přežití 1–2 roky u relabujících/refrakterních pacientů [Sanchorawala, 2007b; Palladini, 2012b]. Nicméně terapie u nemocných s AL amyloidózou je často spojena s významnou renální a kardiální toxicitou a celkovou alterací stavu, proto zejména u nemocných s renální nedostatečností či vysokou proteinurií by měla být léčba vedena velmi obezřetně [Specter, 2011]. Léčba bývá navíc spojena i s významným vzestupem v hodnotách kardiálních biomarkerů, zejména natriuretických peptidů, což mimo jiné činí posouzení srdeční orgánové odpovědi velmi problematické [Tapan, 2010]. Ve studii Moreau et al. bylo užito kombinace lenalidomidu, melfalanu a dexamethasonu u 26 nemocných s nově diagnostikovanou AL amyloidózou. 58 % nemocných dosáhlo hematologické odpovědi, včetně 23 % kompletních remisí [Moreau, 2010]. Některé studie se stejným léčebným režimem však referují o nižším procentu léčebných odpovědí. Práce hodnotící kombinaci lenalidomidu, dexamethaso-nu a cyklofosfamidu v souborech z části předlé-čených pacientů uvádějí přibližně 60 % hematologických odpovědí (5–11 % CR) [Palladini, 2013; Kumar, 2012].
Režimy s lenalidomidem nejsou v současnosti doporučovány v iniciální terapii nemocných s AL amyloidózou, nicméně jsou léčebnou možností pro nemocné refrakterní na režimy s bortezomibem či jiná chemoterapeutika, či pro nemocné s významnou polyneuropatií (úroveň důkazu IIa, stupeň doporučení B). Iniciální dávka je doporučována 15 mg/den. Lék není v ČR v roce 2018 pro léčbu AL amyloidózy registrován a je nutné žádat úhradu.
15.3 Pomalidomid v léčbě AL amyloidózy
Pomalidomid v kombinaci s dexamethasonem byl iniciálně testován ve skupině předléčených nemocných v práci z Mayo Clinic. Míra léčebných odpovědí dosáhla 41 %, v případě IMID-refrakterních nemocných 43 %. Jednoleté přežití dosahovalo 77 % [Dispenzieri, 2007]. Jiná studie hovoří o 50% léčebné odpovědi [Sanchorawala, 2016]. Palladini et al. ve studii s 28 pacienty s relabující/refrakterní AL amyloidózou uvádí 68 % léčebných odpovědí, včetně 29 % VGPR/CR [Palladini, 2012]. Pomalidomid v kombinaci s kortikoidy není doporučován pro iniciální terapii nemocných s AL amyloidózou (úroveň důkazu IIa, stupeň doporučení B). Lze jej však zvážit u relabujících/refrakterních pacientů v dobré klinické kondici. Lék není v ČR v roce 2018 pro léčbu AL amyloidózy registrován a je nutné žádat úhradu.
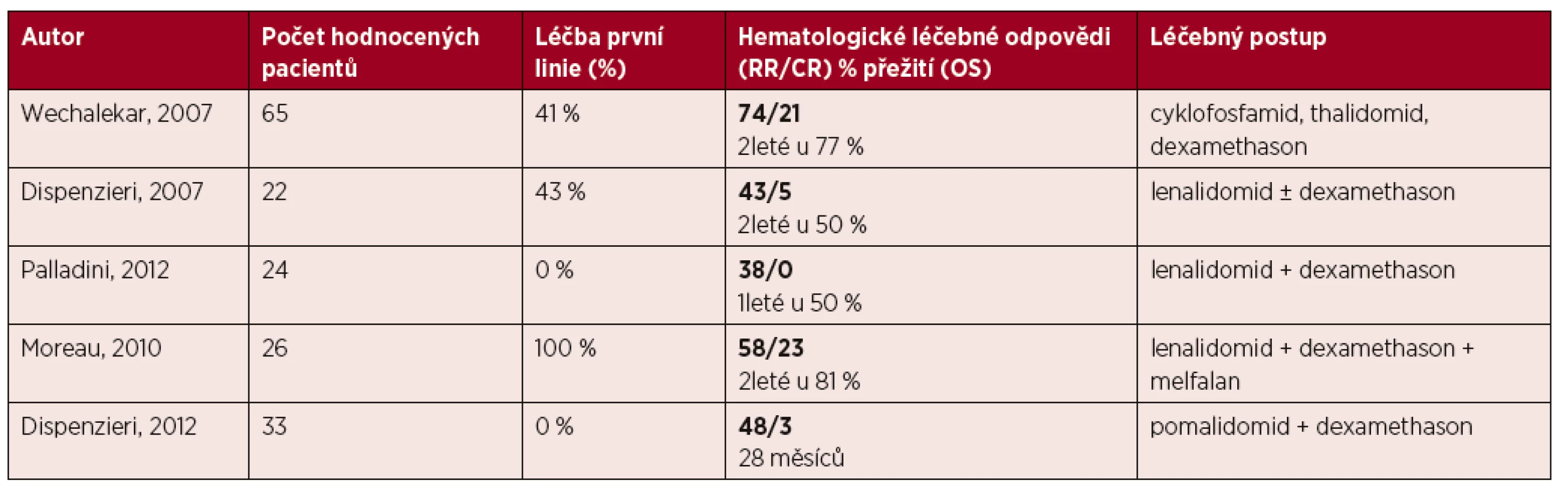
16 INHIBITORY PROTEAZOMU V LÉČBĚ AL AMYLOIDÓZY
16.1 Bortezomib v léčbě AL amyloidózy
16.1.1 Bortezomib v monoterapii a v kombinaci s dexamethasonem
Bortezomib v monoterapii byl prospektivně hodnocen v klinické studii fáze I/II s eskalací dávek a dobrou tolerancí až do dávky 1,6 mg/m2 1krát týdně nebo 1,3 mg/m2 2krát týdně [Comenzo, 2010; Reece, 2011]. Hematologická odpověď byla 39 % (CR 11 %) při nižších dávkách, při dávce 1,6 mg/m2 1krát týdně byla hematologická odpověď 69 % (z toho CR 38 %) a 67 % (z toho CR 24 %) při dávce 1,3 mg/m2 2krát týdně [Comenzo, 2010]. Čas do dosažení léčebné odpovědi byl kratší při dávkovacím schématu 2krát týdně, ovšem dávkovací schéma 1krát týdně bylo lépe tolerováno. Kombinace bortezomibu a dexamethasonu (BDex) je spojena s vysokým procentem hematologických léčebných odpovědí (80–94 %) [Kastritis, 2007]. Retrospektivní studie 94 pacientů s AL amyloidózou udává hematologickou odpověď 71 %, z toho bylo dosaženo 25 % CR, dříve neléčení pacienti dosáhli CR v 47 % případů [Kastritis, 2010]. Medián doby do dosažení hematologické odpovědi byl 1,7 měsíce. Kombinace bortezomibu a dexamethasonu je doporučována v iniciální terapii i léčbě relabujících nemocných s AL amyloidózou.
16.1.2 Kombinovaná schémata s bortezomibem
V současné době jsou ponejvíce v první linii terapie, ale i u relabujících nemocných doporučována kombinovaná schémata bortezomibu, dexamethasonu a alkylačních cytostatik, neboť jsou spojena s nejvyšším procentem nejen hematologických, ale i orgánových odpovědí při akceptovatelné toxicitě (úroveň důkazu IIa, stupeň doporučení B).
Jedna z prvních prací Mikhaela et al. uvádí, že kombinace cyklofosfamidu, bortezomibu a dexamethasonu (CyBorD) vedla k dosažení VGPR/CR u 16 z 17 léčených pacientů [Mikhael, 2012] s minimální toxicitou. I řada dalších prací udává míru hematologické léčebné odpovědi v rozmezí 62–94 % při 17–61 % CR. V současné době největší publikovaný soubor zahrnoval 230 pacientů, kteří obdrželi kombinaci CyBorD a ve kterém bylo dosaženo 62 % hematologických remisí, z čehož 21 % bylo kompletních [Palladini, 2015]. V této práci však byla identifikována skupina nemocných ve stadiu III Mayo klasifikace s hladinami NT-proBNP nad 8500 pg/ml (tedy stadium IIIb) s výrazně krátkým přežitím a ve které byla zaznamenána 66% míra úmrtí v prvních 12 měsících. Ačkoliv 39 % těchto nemocných vykazovalo hematologickou léčebnou odpověď, bohužel nadále byla u těchto pacientů sledována srdeční progrese onemocnění.
Venner et al. porovnával soubor 69 pacientů léčených režimem CyBorD a 69 nemocných léčených režimem CTD. Léčba režimem CyBorD byla spojena s vyšším procentem kompletních remisí (40 vs. 25 %) a delší dobou do progrese (28 vs. 14 měsíců), jednoleté přežití bylo identické pro obě skupiny nemocných (65 vs. 67 %) [Venner, 2014]. Režim CyBorD představuje optimální režim pro indukční terapii před ASCT s ohledem na plánovaný bezpečný a dostatečný sběr periferních kmenových buněk.
V současnosti existuje jediná randomizovaná studie porovnávající kombinaci bortezomibu, melfalanu a dexamethasonu (BMDex) s melfalanem a dexamethasonem (MDex). Režim BMDex byl spojen s rapidnějším efektem terapie, vyšším počtem hematologických odpovědí (81 vs. 56 %) i větší hloubkou odpovědi (CR/VGPR 64 vs. 38 %) a s prodloužením celkového přežití. Míra orgánové léčebné odpovědi byla u hodnocených nemocných stejná, bez rozdílu vyznělo i celkové přežití. Režim Mdex byl naopak spojen s nižší toxicitou. Ve skupině BMDex byla zaznamenána menší míra kardiálních orgánových progresí [Kastritis, 2016]. Režim BMDex je považován za nový standard pro nově diagnostikované pacienty s AL amyloidózou, kteří nejsou referováni k ASCT. Přehled některých klinických studií s bortezomibem u pacientů s AL amyloidózou uvádí tabulka 18.

16.1.3 Nežádoucí účinky léčby
Nežádoucí účinky terapie bortezomibem jsou popisovány 30 % nemocných, nejčastěji se jedná o retenci tekutin, hypotenzi, projevy polyneuropatie periferní či viscerální a gastrointestinální obtíže [Kastritis, 2010]. V současnosti jsou jako u mnohočetného myelomu preferovány režimy s týdenním podáváním bortezomibu. Sidana et al. prokázal podobnou účinnost a nižší míru neuropatií u ALA při subkutánním podání bortezomibu 1krát týdně ve srovnání s jinými schématy režimů na bázi bortezomibu [Sidana,2017]. U fragilních nemocných lze zvážit redukci dávky na 1 mg/m2. Stejně jako v případě mnohočetného myelomu je doporučována profylaxe antivirotiky proti infekci virem varicella zoster.
16.2 Ixazomib a carfilzomib v léčbě AL amyloidózy
Ixazomib představuje novou generaci proteazomového inhibitoru s plně perorálním podáním. Stejně jako v případě mnohočetného myelomu, kde prokázal účinnost zejména v kombinacích, je testován i u AL amyloidózy. Výsledky studie fáze I/II u relabující/refrakterní AL amyloidózy referuje o 52 % hematologických a 56 % orgánových odpovědí při akceptovatelné toxicitě [Sanchorawala, 2017]. V současnosti probíhá studie fáze III a studie fáze I/II v kombinaci s cyklofosfamidem a dexamethasonem. Dávky léku je nutné redukovat při těžší renální insuficienci.
Carfilzomib – jako další z nových typů proteazomových inhibitorů – je rovněž testován i u AL amyloidózy. Cohen et al. hodnotil kombinaci carfilzomibu a dexamethasonu ve skupině 28 nemocných s relabující/refrakterní AL amyloidózou. Hematologické odpovědi bylo dosaženo u 63 % nemocných s 21 % orgánových odpovědí. Nicméně léčba byla spojena s vysokým výskytem závažných komplikací, zejména kardiálních [Cohen, 2016]. Další studie s carfilzomibem v současnosti probíhají.
17 LÉČBA RELABUJÍCÍCH A REFRAKTERNÍCH PACIENTŮ
Tabulka 19 shrnuje doporučení pro léčbu relabujících a refrakterních nemocných. U relabující/refrakterní AL amyloidózy může být léčba opakovaná (zvláště pokud byla dříve účinná, dobře tolerovaná a délka do další progrese je klinicky významná) nebo může být zvážena léčba i jinými preparáty. K dispozici jsou data ze studií fáze II. Mezi léky s efektem u relabujících nemocných patří například bendamustin [Lagos, 2016; Milani, 2018]. V léčbě relapsu je opět preferována léčba v klinických studiích, pokud jsou dostupné [Dispenzieri, 2015; Palladini, 2016].

18 DARATUMUMAB V LÉČBĚ AL AMYLOIDÓZY
Monoklonální anti-CD38 protilátka daratumumab je v současnosti používána u relabujících/refrakterních pacientů s mnohočetným myelomem. Kaufman et al. referuje o výborném efektu terapie ve skupině 25 nemocných s relabující/refrakterní AL amyloidózou, kdy hematologické odpovědi bylo dosaženo u 76 % nemocných, přičemž procento CR dosahovalo 36 %. Terapie byla dobře tolerována i u nemocných se srdečním postižením a medián dosažení léčebné odpovědi představoval jeden měsíc [Kaufman, 2017]. Terapie daratumumabem tedy představuje slibnou terapii pro nemocné s AL amyloidózou, studie fáze II a III v současnosti probíhají.
19 PERSPEKTIVY V LÉČBĚ AL AMYLOIDÓZY
Řada dalších molekul se v současnosti jeví jako perspektivní v terapii AL amyloidózy a jejich účinek je prověřován ve studiích fáze I/II.
Mezi léky interferujícími s formací amyloidových depozit patří antibiotikum doxycyklin. Preklinické studie prokázaly, že doxycyklin dokáže přerušit amyloidové fibrily u AL i transthyretinové amyloidózy. Wechalekar et al. prokázal, že přidání doxycyklinu k terapii u nemocných s kardiální amyloidózou vedlo k redukci časné mortality a vedlo k vyšší míře léčebných odpovědí a delšímu přežití [Wechalekar, 2015b]. Proto by jako součást iniciální terapie a antimikrobní profylaxe měl být preferován doxycyklin (úroveň důkazu IIa, stupeň doporučení B).
Další léky jsou cíleny na amyloidová depozita. Mezi první patřila monoklonální protilátka NEOD001, která se cíleně vázala na protofilamenta a fibrily tvořené lehkými řetězci imunoglobulinu a které byly následně odstraňovány fagocytózou. Iniciální práce fáze I/II hovořila o 60 % orgánových odpovědí [Gertz, 2016]. Nicméně na jaře roku 2018 byly další vývoj a klinické zkoušení protilátky pro nedostatečný efekt zastaveny.
Jiný princip představuje léčba cílená proti sérové amyloidové P komponentě (SAP), proteinu, jenž je součástí všech typů amyloidových depozit a chrání amyloidová depozita proti degradaci. Pepys et al. prokázal, že molekula R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid (CPHPC, miridesap) dokáže redukovat cirkulující hladiny SAP a následná aplikace anti-SAP monoklonální protilátky vede k indukci makrofágové odpovědi a odstraňování tkáňových amyloidových depozit [Bodin, 2010]. Pilotní studie prokázala překvapivé výsledky a v současnosti probíhají klinická hodnocení u léčených nemocných i u nemocných po proběhlé terapii [Richards, 2015].
20 TRANSPLANTACE SRDCE, PŘÍPADNĚ JINÝCH ORGÁNŮ, NÁSLEDOVANÁ VYSOKODÁVKOVANOU CHEMOTERAPIÍ S AUTOLOGNÍ TRANSPLANTACÍ KRVETVORNÝCH BUNĚK
Těžce poškozené srdce je kontraindikací vysokodávkované chemoterapie. Ale i konvenční léčba obsahující vysoké dávky dexametazonu, která obvykle způsobuje retenci tekutin, nemusí být proveditelná při závažném poškození srdce. Pokud jde o mladšího nemocného a ostatní orgány nejsou kriticky poškozeny, je možné zvážit iniciální transplantaci srdce (úroveň důkazu IV, stupeň doporučení C). Tím se výrazně zlepší celková zdatnost a pak je možné použít vysokodávkovanou chemoterapii s podporou ASCT s cílem eradikovat patologický klon plazmocytů. V literatuře lze nalézt četné popisy menších skupin nemocných, které dokládají přínos provedení orgánové transplantace nejvíce poškozeného orgánu (tedy nejen srdce) následované léčbou amyloidózy [Adam, 2013; Gatt, 2013]. Podstatné je, aby pacient byl v celkově dobrém stavu a měl amyloidózou závažně poškozen pouze jeden orgán, který je nahrazen orgánem transplantovaným. Zřejmě však i do budoucna půjde spíše o výjimečnou léčbu [Adam, 2013].
U vybraných nemocných s izolovaným renálním postižením a celkově limitovaným onemocněním lze zvážit provedení transplantace ledviny (úroveň důkazu IV, stupeň doporučení C). Provedení transplantace ledviny jako náhrady vlastního selhávajícího orgánu je doporučováno po dosažení kompletní hematologické remise onemocnění, nejčastěji v návaznosti na proběhlou ASCT. 5leté přežití nemocných je udáváno v rozmezí 67–78 % [Dispenzieri, 2015].
Transplantace jater v případě AL amyloidózy není doporučována, prognóza nemocných je krajně nepříznivá, pětileté přežití je udáváno ve 22 % (úroveň důkazu IV, stupeň doporučení C) [Dispenzieri, 2015].
21 PODPŮRNÁ PÉČE PŘI LÉČBĚ AL AMYLOIDÓZY
Systémová AL amyloidóza je nejen hematologické, ale zejména multiorgánové postižení a nemocní jsou zpravidla postiženi zhoršením či selháním funkcí jednoho a více tělesných orgánů. Nejen v diagnostice, ale i během vedení terapie je nutná spolupráce s odborníky z jiných oborů. Nejčastější spolupracující specialisté jsou z oboru kardiologie, nefrologie, neurologie, nutriční péče a mnoho dalších. Je více než vhodné, aby při specializovaných centrech působil multidisciplinární tým lékařů se zaměřením na problematiku amyloidóz, neboť, jak bylo výše řečeno, amyloidózy představují relativně vzácnou skupinu onemocnění a bylo by vhodné péči o pacienty svěřit do rukou odborníků.
Tabulka 20 částečně shrnuje specifickou farmakologickou a nefarmakologickou podpůrnou péči o nemocné s AL amyloidózou [Dispenzieri, 2015; Palladini, 2016].
![Podpůrná terapie u nemocných s AL amyloidózou (upraveno
podle [Dispenzieri, 2015; Palladini, 2016])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/7c568194f2d088ed181753fb6d581afe.png)
22 DOPORUČENÍ PRO LÉČBU AL AMYLOIDÓZY
- Principem léčby je eliminace patologického klonu se zastavením produkce amyloidogenních lehkých řetězců, redukcí jejich sérových hladin s cílem dosažení alespoň velmi dobré parciální hematologické remise (úroveň důkazu IIa, stupeň doporučení B).
- Dosažení hematologické léčebné odpovědi je podmínkou pro zlepšení funkce postižených orgánů a dosažení tzv. orgánové léčebné odpovědi (úroveň důkazu IIa, stupeň doporučení B).
- Léčba je volena individuálně s využitím risk-adapted strategie (úroveň důkazu IV, stupeň doporučení C).
- Pouze část nemocných s AL amyloidózou splňující výběrová kritéria je možné bezpečně léčit vysokodávkovanou terapií s podporou autologního štěpu, tito nemocní s nízkou mírou orgánového postižení z této terapie profitují (úroveň důkazu IIa, stupeň doporučení B).
- Léčba kombinací melfalanu a dexamethasonu je možnou variantou pro nemocné, kteří nejsou vhodnými kandidáty vysokodávkované terapie (úroveň důkazu IIa, stupeň doporučení B).
- Kombinovaná schémata s bortezomibem jsou vhodnými léčebnými režimy pro všechny nově diagnostikované nemocné bez ohledu na to, zda jsou či nejsou vhodnými kandidáty vysokodávkované terapie (úroveň důkazu IIa, stupeň doporučení B). Kombinovaná schémata s bortezomibem jsou vhodnými léčebnými režimy pro relabující nemocné (úroveň důkazu IIa, stupeň doporučení B).
- Schémata s imunomodulačními látkami jsou indikována u relabujících/refrakterních nemocných (úroveň důkazu IIa, stupeň doporučení B).
- Multioborová spolupráce při péči o nemocné s AL amyloidózou je naprosto nezbytná.
Seznam použitých zkratek
AF – alkalická fosfatáza
AI – apolipoprotein
AL – amyloid tvořený lehkými řetězci imunoglobulinu
ALA – AL amyloidóza
ALP – alkalická fosfatáza
anti-CD38 – monoklonální protilátka proti receptoru CD38
A-PC – klonální plazmocyty
aPTT – aktivovaný parciální tromboplastinový čas
ASCT – autologní transplantace krvetvorných buněk
ATTR – transthyretinová hereditární amyloidóza
AV – atrioventrikulární blokáda
Aβ2M – hemodialyzační amyloidóza
BDex – bortezomib, dexamethason
BMDex – bortezomib, melfalan, dexamethason
BNP – mozkový natriumuretický peptid, brain natriuretic peptide
CB-U – proteinurie
CMG – Česká myelomová skupina
CNS – centrální nervová soustava
CPHPC – R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid; miridesap
CR – kompletní remise
CT – počítačová tomografie
CyBorD – cyklofosfamid, bortezomib, dexamethason
dFLC – diference hladin volných lehkých řetězců imunoglobulinu
DKK – dolní končetiny
DNA – deoxyribonukleová kyselina
DPD scan – scintigrafie s užitím kyseliny 3,3-difosfono-1,2-propanodikarboxylové
eGRF – clearance kreatininu
ECHO – echokardiografie
EKG – elektrokardiografie
EMB – endomyokardiální biopsie
EMG – elektromyografie
ESRD – end-stage renal disease
FFPE – materiál fixovaný formalinem a zalitý do parafinu
GF – glomerulární filtrace
GIT – gastrointestinální trakt
HCDD – nemoc z ukládání těžkých řetězců
HRCT – počítačová tomografie s vysokým rozlišením
IFE – imunofixační elektroforéza
IgA – typ monoklonálního imunoglobulinu A
IgD – typ monoklonálního imunoglobulinu D
IgG – typ monoklonálního imunoglobulinu G
IgM – typ monoklonálního imunoglobulinu M
IHC – imunohistochemie
IMIdy – imunomodulační léky
IMWG – International Myeloma Working Group
KD – kostní dřeň
LCDD – nemoc z ukládání lehkých řetězců
LŘ – lehké řetězce (v séru)
MDex – melfalan, dexamethason
M-FC – multiparametrická průtoková cytometrie
MGRS – monoclonal gammopathy of renal signifikance; monoklonální gamapatie renálního významu
MGUS – monoklonální gamapatie nejistého významu
MIg – monoklonální imunoglobulin
MM – mnohočetný myelom
MR – magnetická rezonance
NGS – sekvenování nové generace
NHL – non-Hodgkinův lymfom
N-PC – polyklonální plazmocyty
NR – bez léčebné odpovědi
NS – nefrotický syndrom
NT-proBNP – propeptid mozkového natriuretického peptidu
OS – celkové přežití
PC – plazmocyty
PCR – polymerázová řetězová reakce
PET/CT – pozitronová emisní tomografie/počítačová tomografie
PR – parciální remise
RR – počet léčebných odpovědí
RTG – standardní radiografie
SAA – sérový amyloid A
SAP – glykoprotein (sérová amyloidní P komponenta)
SPE – standardní elektroforéza bílkovin séra
sy – syndrom
TK – tlak krve
TnI – troponin I
TnT – troponin T
TRM – riziko úmrtí v souvislosti s transplantací
TTR – transthyretin
UPE – elektroforéza proteinů moči
UZ – ultrazvukové vyšetření
VFN – Všeobecná fakultní nemocnice v Praze
VGPR – velmi dobrá parciální remise
VLŘ – volné lehké řetězce
WM – Waldenströmova makrogobulinemie
Podíl autorů na rukopisu
T. P. (vytvoření základní osnovy guidelines, napsání rukopisu, revize rukopisu); R. H. (vytvoření základní osnovy, napsání dílčí části rukopisu, revize rukopisu); Z. A. (připomínky, revize rukopisu); P. F. (napsání dílčí části rukopisu, připomínky, revize rukopisu); P. F. (napsání dílčí části rukopisu, připomínky, revize rukopisu); D. H. (připomínky, revize rukopisu); T. J. (připomínky, revize rukopisu); Z. Ch. (připomínky, revize rukopisu); V. M. (připomínky, revize rukopisu); R. R. (připomínky, revize rukopisu); V. Š. (připomínky, revize rukopisu); I. Š. (připomínky, revize rukopisu)
Prohlášení o konfliktu zájmů
R. H. (honoráře, přednášky, konzultace a nefinanční podpora od firmy Janssen); T. J. (honoráře, přednášky: Amgen, Takeda, Janssen); V. M. (honoráře, přednášky, konzultace a nefinanční podpora od firmy Janssen); T. P.; Z. A.; P. F.; P. F.; D. H.; Z. Ch.; R. R.; V. Š. a I. Š. (honoráře: Celgene, Amgen, Janssen-Cilag, Takeda, BMS, Novartis, Sanofi; přednášky: Celgene, Amgen, Janssen-Cilag, Takeda, BMS, Novartis; konzultace Celgene, Amgen, Janssen-Cilag, Takeda, BMS, Novartis, Sanofi; nefinanční podpora Celgene, Amgen, Janssen-Cilag, BMS) nedeklarují v souvislosti se vznikem a přípravou rukopisu žádný konflikt zájmů.
Doručeno do redakce: 24. 1. 2019.
Přijato po recenzi: 28. 1. 2019.
KORESPONDENČNÍ ADRESA:
MUDr. Tomáš Pika, Ph.D.
Hemato-onkologická klinika, LF UP a FN Olomouc
I. P. Pavlova 185/6, 779 00 Olomouc
e-mail: tomas.pika@seznam.cz
Zdroje
Použitá literatura pro diagnostickou část
1. Adam Z, Ščudla V. Klinické projevy a diagnostika AL amyloidózy a některých dalších typů amyloidóz. Vnitř. Lék. 2001; 47 : 36–45.
2. Adam Z, Elleder M, Moulis M, et al. Přínos PET-CT vyšetření pro rozhodování o léčbě lokalizované nodulární formy plicní AL-amyloidózy. Vnitř. Lék. 2012; 58 : 241–252. (Adam 2012a)
3. Adam Z, Stork M, Pour L, et al. Výsledky léčby AL-amyloidózy léčebnými režimy obsahujícími bortezomib, dexametazon a dále cyklofosfamid anebo doxorubicin. Vnitř. Lék. 2012; 58 : 896–903. (Adam 2012b)
4. Alpers CE, Cotran RS. Neoplasia and glomerular injury. Kidney Int 1986; 30 : 465–470.
5. Arbustini E, Verga L, Concardi M, et al. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid 2002; 9 : 108–114.
6. Bijzet J, Van Gameren II, Muller Kobold AC, et al. Heavy/light chain analysis can replace IFE in an algorithm utilizing Free light chain and urinary protein electrophoresis for identification of AL amyloidosis patients. Presented at XIII International Symposium on Amyloidosis 2012.
7. Bird J. Guidelines on the diagnosis and management of AL amyloidosis. Br J Haematol 2004; 125 : 681–700.
8. Bohle A, Wehrmann R, Eissele R, et al. The long-term prognosis of AA and AL renal amyloidosis and the pathogenesis of chronic renal failure in renal amyloidosis. Path Res Pract 1993; 189 : 316–321.
9. Bochtler T, Hegenbart U, Kunz C, et al. Gain of chromosome 1q21 is an independent adverse prognostic factor in light chain amyloidosis patients treated with melphalan/dexamethasone. Aamyloid 2014; 21(1): 9–17.
10. Bochtler T, Hegenbart U, Kunz Ch, et al. Translocation t(11; 14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol 2015; 33 : 1371–1378.
11. Bochtler T, Hegenbart U, Kunz Ch, et al. Prognostic impact of cytogenetic aberrations in AL amyloidosis after high-dose melphalan: a long-term follow up. Blood 2016; 128(4): 594–602.
12. Bradwell AR. Serum free light chain analysis (plus Hevylite) 7th. ed. Birmingham: The Binding Site Ltd, 2015; 296–314.
13. Chyra Kufova Z, Sevcikova T, Januska J, et al. Newly designed 11-gene panel reveals first case of hereditary amyloidosis captured by massive parallel sequencing. J Clin Pathol 2018; 71(8): 687–694.
14. Collins A, Smith R, Stone J. Classification of amyloid deposits in diagnostic cardiac specimens by immunofluorescence. Cardiovasc Pathol 2009; 18 : 205–216.
15. Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia 2012; 26 : 2317–2325.
16. Comenzo RL, Wally J, Kica G, et al. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: association with dominant amyloid-related organ involvement and survival after stem cell transplantation. Br J Haematol 1999; 106 : 744–751.
17. Dember LM. Renal amyloidosis. In: Gertz MA, Rajkumar SV. Amyloidosis: diagnosis and treatment, 1st ed. New York: Springer Science 2010; 129–143.
18. Dinner S, Witteles W, Witteles R, et al. M. The prognostic value of diagnosing concurrent multiple myeloma in immunoglobulin light chain amyloidosis. Br J Haematol 2013; 161 : 367–372.
19. Dispenzieri A, Kyle RA, Gertz MA, et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet 2003; 361 : 1787–1789.
20. Dispenzieri A, Gertz MA, Kyle RA, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patiens with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2004; 104 : 1881–1887. (Dispenzieri 2004a)
21. Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: A staging system for primary systemic amyloidosis. J Clin Oncol 2004; 22 : 3751–3757. (Dispenzieri 2004b)
22. Dispenzieri A, Lacy MQ, Katzmann JA, et al. Absolute values of imunoglobulin free light chains are prognostic in patiens with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2006; 107 : 3378–3383.
23. Dispenzieri A. Assessing response and prognosis in AL amyloidosis. In. Gertz MA, Rajkumar SV. Amyloidosis: diagnosis and treatment, 1st ed. New York: Springer Science 2010, 77–93. (Dispenzieri 2010a)
24. Dispenzieri A, Dingli D, Kumar SK, et al. Discordance between serum cardiac biomarker and immunoglobulin-free light-chain amyloidosis treated with immune modulatory drugs. Am J Hematol 2010; 85 : 757–759. (Dispenzieri 2010b)
25. Dispenzieri A, Gertz MA, Buadi F. What do I need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev 2012; 26 : 137–154.
26. Dittrich T, Bochtler T, Kimmich Ch, et al. AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have excellent prognosis. Blood 2017; 130(5): 632–642.
27. D’Souza A, Theis J, Quint P, et al. Exploring the amyloid proteome in immunoglobulin-derived lymph node amyloidosis using laser microdissection/tandem mass spectrometry. Am J Hematol 2013; 88 : 577–580.
28. Dogan A. Amyloidosis: Insights from Proteomics. Annual Review of Pathology: Mechanisms of Disease 2017; 12(1): 277–304.
29. Esplin BL, Gertz MA. Current trends in diagnosis and management of cardiac amyloidosis. Curr Probl Cardiol 2013; 38 : 53–96.
30. Falk RH, Comenzo RL, Skinner M. The systemic amyloidosis. New Engl J Med 1997; 337 : 898–909.
31. Fielder K, Durie BGM. Primary amyloidosis associated with multiple myeloma. Am J Med 1986; 80 : 413–418.
32. Fikrle M, Paleček T, Kuchyňka P, et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa 2013; 55(1): e60–75.
33. Fikrle M, Paleček T, Mašek M, et al. The diagnostic performance of cardiac magnetic resonance in detection of myocardial involvement in AL amyloidosis. Clin Physiol Funct Imaging 2016; 36 : 218–224.
34. Georgiades CS, Neyman EG, Barish MA, et al. Amyloidosis: review and CT manifestation. Radiographics 2004; 24 : 405–416.
35. Gertz MA, Kyle RA, Greipp PR. The plasma cell labeling index: a valuable tool in primary systemic amyloidosis. Blood 1989; 74 : 1108–1111.
36. Gertz MA, Kyle RA, Greipp PR, et al. Beta2-microglobulin predicts survival in primary systemic amyloidosis. Am J Med 1990; 89 : 609–614.
37. Gertz MA, Greipp PR, Kyle RA. Classification of amyloidosis by the detection of clonal exces of plasma cells in the bone marrow. J Lab Clin Med 1991; 118 : 33–39.
38. Gertz MA, Kyle RA. Amyloidosis: prognosis and treatment. Semin Arthritis Rheum 1994; 24 : 124–138.
39. Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis: recognition, confirmation, prognosis, and therapy. Mayo Clin Proc 1999; 74 : 490–494.
40. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International symposium on amyloid and amyloidosis. Am J Hematol 2005; 79 : 319–328.
41. Gertz MA, Lacy MQ, Dispenzieri A, et al. Transplantation for amyloidosis. Curr Opin Oncol 2007; 19 : 136–141.
42. Gertz MA, Hayman SR. Immunoglobulin light chain amyloidosis. In: Rajkumar SV, Kyle RA. Treatment of multiple myeloma and related disorders. Cambridge: Cambridge University Press 2009; 112–128.
43. Gertz MA, Rajkumar SV. Amyloidosis, contemporary hematology. Springer Science+Business Media, LLC 2010; 33–48.
44. Gertz MA et Merlini G. Definition of organ involvement and response to treatment in AL amyloidosis: an updated consensus opinion. Amyloid 2010; 17 : 48–49.
45. Gertz MA, Buadi FK, Hayman SR, et al. Immunoglobulin D amyloidosis: a distinct entity. Blood 2012; 119(1): 44–48.
46. Gertz MA. Immunoglobulin light chain amyloidosis: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol 2011; 86 : 181–186.
47. Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol 2016; 91 : 948–956.
48. Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on diagnosis and investigation of AL amyloidosis. Brit J Hematol 2015; 168 : 207–218.
49. Hassan W, Al-Sergani H, Mourad W, et al. Amyloid heart dinase. Tex Heart Inst J 2005; 32 : 178–184.
50. Hawkins PN. Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Curr Opin Nephrol Hypertens 2002; 11 : 649–655.
51. Hosch W, Kristen AV, Libicher M, et al. Late enhancement in cardiac amyloidosis: correlation of MRI enhancement pattern with histopatological findings. Amyloid 2008; 15 : 196–204.
52. Howie AJ, Brewer DB, Howell D, et al. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest 2008; 88 : 232–242.
53. Jelinek T, Bezdekova R, Zatopkova M, et al. Current applications of multiparameter flow cytometry in plasma cell disorders. Blood Cancer J 2017; 7: e617.
54. Jelinek T, Kryukova E, Kufova Z, Kryukov F, Hajek R. Proteasome inhibitors in AL amyloidosis: focus on mechanism of action and clinical activity. Hematol Oncol 2017; 35(4): 408–419.
55. Jelinek T, Kufova Z, Hajek R. Immunomodulatory drugs in AL amyloidosis. Crit Rev Oncol Hematol 2016; 99 : 249–260.
56. Kapoor P, Thenappan T, Singh E, et al. Cardiac amyloidosis: A practical approach to diagnosis and management. Am J Med 2011; 124 : 1006–1015.
57. Katzmann JA, Abraham RS, Dispenzieri A, et al. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem 2005; 51 : 878–881.
58. Kovářová L, Varmužová T, Žarbochová P, et al. Flow cytometry in monoclonal gammopathies. Klin Onkol 2011; 24(Suppl): S24–S29.
59. Koyama J, Falk RH. Prognostic significance of strain doppler imaging in light-chain amyloidosis. J Am Coll Cardiol Img 2010; 3 : 333–342.
60. Krejčí K, Zadražil J, Tichý T. Nefropatie v ultrazvukovém a histologickém obraze. 1. vydání. Praha: Maxdorf Jessenius 2011; 186.
61. Kristen AV, Giannitsis E, Lehrke S, et al. Assessment of disease severity and outcome in patients with systemic light-chain amyloidosis by the high-sensitivity troponin T assay. Blood 2010; 116 : 2455–2461.
62. Kumar S, Dispenzieri A, Lacy MQ, et al. Improved survival in light chain amyloidosis. Amyloid 2010; 17(Suppl 1): 89–95.
63. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30 : 989–995.
64. Kyle RA, Greipp PR, O´Fallon WM. Primary systemic amyloidosis: multivariate analysis for prognostic factors in 168 cases. Blood 1986; 68 : 220–224.
65. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted county, Minessota,1950 through 1989. Blood 1992; 79 : 1817–1822.
66. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical features in 474 and laboratory cases. Semin Hematol 1995; 32 : 45–59. (Kyle 1995a)
67. Kyle RA. Multiple myeloma and other plasma cell disorders. In: Hofman R, Bentz EJ Jr, Shattil SJ, et al. Hematology: basic principles and practice, 2nd ed. New York: Churchill Livingstone 1995; 1354–1374. (Kyle 1995b)
68. Kyle RA, Gertz MA, Greip PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. New Engl J Med 1997; 336 : 1202–1207.
69. Kyle RA, Durie BG, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. 2010; 24(6): 1121–1127.
70. Kyriakides T, Marques B, Panousopoulos A, et al. Amyloid myopathy: evidence for mechanical injury to the sarcolemma. Clin Neuropathol 2002; 21 : 145–148.
71. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. New England J Med 2002; 346 : 1786–1791.
72. Lachmann HJ, Gallimore R, Gillmore JD, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol 2003; 122 : 78–84.
73. Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal signifikance (MGRS): when MGUS is no longer undetermined or insignificant. Blood 2012; 120(22): 4292–4295.
74. Loo D, Mollee PN, Renaut P, Hill MM. Proteomics in molecular diagnosis: typing of amyloidosis. J Biomed Biotechnol 2011; 2011 : 754109.
75. Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005; 111 : 186–193.
76. Maceira AM, Prasad SK, Hawkins PN, et al. Cardiovascular magnetic resonance and prognosis in cardiac amyloidosis. J Cardiovasc Magn Reson 2008; 10 : 54–59.
77. Madan S, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patiens with previous diagnosis of multiple myeloma. Mayo Clin Proc 2010; 85 : 232–236.
78. Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annual Review of Biochemistry 2001; 70(1): 437–473.
79. Maisnar V, Tichý M, et al. Monoklonální imunoglobuliny – výskyt, význam a možnosti jejich průkazu. 1. vydání. Hradec Králové: Nucleus 2012; 125.
80. Marcus A, Sadimin E, Richardson M, et al. Fluorescence microscopy is superior to polarized microscopy for detecting amyloid deposits in Congo red-stained trephine bone marrow biopsy specimens. Am J Clin Pathol 2012; 138 : 590–593.
81. Merlini G. AL amyloidosis: diagnosis and prognosis. Haematologica 2007; 92(Suppl 2): 58–59.
82. Milani P, Basset M, Russo, et al. Patients with light-chain amyloidosis and low free light-chain burden have distinct clinical features and outcome. Blood 2017; 130(5): 625–631.
83. Mumford AD, O´Donnell J, Gillmore JD. Bleeding symptoms and coagulation abnormalities in 337 patients with amyloidosis. Br J Haematol 2000; 110 : 454–460.
84. Murtagh B, Hammill SC, Gertz MA, et al. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol 2005; 95 : 535–537.
85. Nasr SH, Galgano SJ, Markowitz GS, et al. Immunofluorescence on pronase-digested paraffin sections: a valuable salvage technique for renal biopsies. Kidney Int 2006; 70 : 2148–2151.
86. Paiva B, Vídriales MB, Pérez JJ, et al. The clinical utility and prognostic value of multiparameter flow cytometry immunophenotyping in light-chain amyloidosis. Blood 2011; 117 : 3613–3616.
87. Palecek T, Fikrle M, Nemecek E, et al. Contemporary treatment of amyloid heart disease. Curr Pharm Des 2015; 21 : 491–506.
88. Palladini G, Malamani G, Co F, et al. Holter monitoring in AL amyloidosis: prognostic implications. Pacing Clin Electrophysiol 2001; 24 : 1228–1233.
89. Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003; 107 : 2440–2445.
90. Palladini G, Lavatelli F, Russo P, et al. Circulating amyloidogenic free light chaos and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood 2006; 107 : 3854–3858.
91. Palladini G, Perlini S, Merlini G. Imaging of systemic amyloidosis. In: Gertz MA, Rajkumar SV: Amyloidosis: diagnosis and treatment. Springer Science and Business Media, LLC 2010; 15–32. (Palladini 2010a)
92. Palladini G, Gertz MA, Kumar S, et al. Validation of the criteria of response to treatment in AL amyloidosis. Blood 2010; 116 : 1364a. (Palladini 2010b)
93. Palladini G, Foli A, Milani P, et al. Best use of cardiac biomarkers in patients with AL amyloidosis and renal failure. Am J Hematol 2012; 87 : 465–471.
94. Palladini G, Barassi A, Klersy C, et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predict survival in AL amyloidosis. Blood 2010; 116 : 3426–3430. (Palladini 2010c)
95. Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005; 46 : 1076–1084.
96. Pepys M. Amyloidosis. Ann Rev Med 2006; 57 : 223–241.
97. Pika T, Hegenbart U, Flodrova P, Maier B, Kimmich C, Schönland SO. First report of ibrutinib in IgM-related amyloidosis: few responses, poor tolerability, and short survival. Blood 2018; 131(3): 368–371.
98. Pika T, Vymětal J, Metelka R, et al. Postižení srdce při AL amyloidóze. Interní Med 2008; 10 : 466–469.
99. Piper C, Butz T, Farr M, Faber L, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue doppler echocardiography, and myocardial biopsy. Amyloid 2010; 17 : 1–9.
100. Quarta CC, Kruger JL, Falk RH. Cardiac amyloidosis. Circulation 2012; 126 : 178–182.
101. Rajkumar SV, Gertz MA, Kyle RA. Primary systemic amyloidosis with delayed progression to multiple myeloma. Cancer 1998; 82 : 1501–1505.
102. Rajkumar SV, Gertz MA. Advances in the treatment of amyloidosis. New Engl J Med 2007; 356 : 2413–2415.
103. Rajkumar SV. Multiple myeloma: 2011 update to diagnosis, risk stratification, and management. Am J Hematol 2011; 86 : 57–62.
104. Rapezzi C, Quarta CC, Guidalotti PL, et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. J Am Coll Cardiol Img 2011; 4 : 659–670.
105. Reyners AKL, Hazenberg BPC, Reitsma WD, et al. Heart rate variability as a predictor of mortality in patients with AA and AL amyloidosis. Eur Heart J 2002; 23 : 157–161.
106. Ruberg FL, Appelbaum E, Davidoff R, et al. Diagnostic and prognostic utility of cardiovascular magnetic resonance imaging in light-chain cardiac amyloidosis. Am J Cardiol 2009; 103 : 544–549.
107. Ryšavá R. Systémové amyloidózy a jejich léčba. Praha: Maxdorf Jessenius 2013; 124.
108. Sanchorawala V, Skinner M, Quillen K, et al. Long-term outcome of patiens with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood 2007; 110 : 3561–3563.
109. Seldin DC, Sanchorawala V. Adapting to AL amyloidosis. Haematologica 2006; 91 : 1591–1595.
110. Schönland SO, Hegenbart U, Bochtler T, et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 2012; 119 : 488–493.
111. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016; 23(4): 209–213.
112. Siragusa S, Morice W, Gertz MA, et al. Asymptomatic immunoglobulin light chain amyloidosis (AL) at the time of diagnostic bone marrow biopsy in newly diagnosed patients with multiple myeloma and smoldering myeloma. A series of 144 cases and review of the literature. Ann Hematol 2011; 90 : 101–106.
113. Snanoudj R, Durrbach A, Gauthier E, et al. Changes in renal function in patients with familial amyloid polyneuropathy treated with orthtopic liver transplantation. Nephrol Dial Transplant 2004; 19 : 1779–1785.
114. Steusloff K, Röcken C, Saeger W. Basement membrane proteins and apolipoprotein E in growth hormone secreting adenomas and their correlation to amyloid: an immunoelectron microscopic study. Endocr Pathol 2000; 11 : 49–56.
115. Ščudla V, Pika T. Současné možnosti diagnostiky a léčby systémové AL-amyloidózy. Vnitř. Lék. 2009; 55(Suppl 1): 77–87.
116. Ščudla V, Minařík J, Pika T. Nemoc z ukládání lehkých řetězců imunoglobulinu (light chain deposition disease). Vnitř. Lék. 2012; 58 : 38–43.
117. Theis JD, Dasari S, Vrana JA, et al. Shotgun-proteomics-based clinical testing for diagnosis and classification of amyloidosis. J Mass Spectrometry 2013; 48(10): 1067–1077.
118. Vacek Z. Histologie a histologická technika. Praha: Avicenum 1988; 376–378.
119. Vávrová J, Tichý M, Maisnar V, et al. Stanovení volných lehkých řetězců a Hevylite u monoklonálních gamapatií. In: Maisnar V, Tichý M, a kol. Monoklonální imunoglobuliny – výskyt, význam a možnosti jejich průkazu. 1. vydání. Hradec Králové: Nucleus 2012; 55–60.
120. Vrana J, Gamez J, Madden B, et al.Classification of amyloidosis by laser microdissection and mass spectrometrybased proteomic analysis in clinical biopsy specimens. Blood 2009; 114 : 4957–4959.
121. Vrana J, Theis J, Dasari S, et al. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometrybased proteomics. Haematologica 2014; 99 : 1239–1247.
122. Warsame R, Kumar SK, Gertz MA, et al. Abnormal FISH in patients with immunoglobulin light chain amyloidosis is a risk factor for cardiac involvement and death. Blood Cancer J 2015; 5: e310.
123. Wechalekar AD, Lachmann HL, Goodman JB, et al. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood 2008; 112 : 40009–40016.
124. Wechalekar AD, Offer M, Gillmore JD, et al. Cardiac amyloidosis, a monoclonal gammopathy and a potentially misleading mutation. Nat Clin Pract Cardiovasc Med 2009; 6 : 128–133.
125. Wechalekar A, Faint J, Bradwell S, et al. Significance of abnormal serum immunoglobulin heavy/light chain ratios (Hevylite) in 294 patients with systemic AL amyloidosis. Haematologica 2011; 96 : 392–398.
126. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 2013; 121 : 3420–3427.
Literatura pro léčebnou část
1. Adam Z, Ščudla V, Krejčí M, et al. Léčba AL amyloidózy v roce 2012, přínos nových léků (bortezomibu, thalidomidu a lenalidomidu). Přehled publikovaných klinických studií. Vnitř. Lék. 2013; 59 : 37–58.
2. Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2010; 468(7320): 93–97.
3. Cibeira MT, Sanchorawala V, Seldin DC, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood 2011; 118 : 4346–4352.
4. Cohen AD, Landau H, Scott EC, et al. Safety and efficacy of carfilzomib (CFZ) in previously-treated systemic light-chain (AL) amyloidosis. Blood 2016; 128 : 645.
5. Comenzo RL, Vosburgh E, Falk RH, et al. Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light-chain) amyloidosis: survival and responses in 25 patients. Blood 1998; 91 : 3662–3670.
6. Comenzo RL, Hegenbart U, Sanchorawala W, et al. High rates of overall and complete haematologic response in a prospective phase ½ study of weeky and twice weekly bortezomib in relapsed AL amyloidosis. Amyloid 2010; 17 : 83–84.
7. Comenzo RL, Reece D, Palladini G, et al. Consensus guidelins for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia 2012; 26 : 2317–2325.
8. Dhopdapkar M, Jagannath S, Vesole D, et al. Treatment of AL-amyloidosis with dexamethasone plus alpha interferon. Leukemia and Lymphoma 1997; 27 : 351–356.
9. Dietrich S, Schönland SO, Benner A, et al. Treatment with intravenous melphalan and dexamethasone is not able to overcome the poor prognosis of patients with newly diagnosed systemic light chain amyloidosis and severe cardiac involvement. Blood 2010; 116(4): 522–528.
10. Dispenzieri A, Lacy M, Rajkumar S, et al. Poor tolerance to high doses of thalidomide in patients with primary systemic amyloidosis. Amyloid 2003; 10 : 257–261.
11. Dispenzieri A, Buadi F, Kumar SK, et al. Treatment of immunoglobulin light chain amyloidosis: Mayo stratification of myeloma and risk-adapted (mSMART) consensus statement. Mayo Clin Proc 2015; 90(8): 1054–1081.
12. Dispenzieri A, Lacy M, Zeldenrust S, et al. The activity of lenalidomide with or without dexamethasone in patiens with primary systemic amyloidosis. Blood 2007; 109 : 465–470.
13. D’Souza A, Dispenzieri A, Wirk B, et al. Improved outcomes after autologous hematopoietic cell transplantation for light chain amyloidosis: A center for international blood and marrow transplant research study. J Clin Oncol 2015; 33(32): 3741–3749.
14. Gavriatopoulou M, Musto P, Caers J, et al. European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia 2018; 32(9): 1883–1898.
15. Gertz M, Lacy M, Dispenzieri A, et al. Risk-adjusted manipulation of melphalan dose before stem cell transplantation in patients with amyloidosis is associated with a lower response rate. Bone Marrow Transplant 2004; 34 : 1025–1031.
16. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis. A consensus opinion from the 10th international symposium on amyloid and amyloidosis. Amer J Hematol 2005; 79 : 319–328.
17. Gertz MA, Lacy MQ, Dispenzieri A, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica 2007; 92(10): 1415–1418.
18. Gertz MA. I dont know how to treat amyloidosis. Blood 2010; 116 : 507–508. (Gertz 2010a)
19. Gertz MA, Merlini G. Definition of organ involvement and response to treatment in AL amyloidosis: an updated consensus opinion. Amyloid 2010; 17(Suppl 1): 48–49. (Gertz 2010b)
20. Gertz MA, Landau H, Comenzo RL, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol 2016; 34(10): 1097–1103.
21. Hájek R, Maisnar V, Krejčí M, Minařík J, Radocha J, et al. Diagnostika a léčba mnohočetného myelomu. Transfuze Hematol dnes 2018; 24(Suppl 1): 1–157.
22. International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies, multiple myeloma and releated disorders: a report of the International Myeloma Working Group. Brit J Haematol 2003; 121 : 749–757.
23. Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007; 357 : 83–93.
24. Kastritis E, Anagnostopoulos A, Roussou M, et al.Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica 2007; 92 : 1351–1358.
25. Kastritis E, Wechalekar E, Dimopoulos M, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. Journal of Clin Oncol 2010; 28 : 1031–1037.
26. Kastritis E, Leleu X, Arnulf B, et al. A randomized phase III trial of melphalan and dexamethasone (MDex) versus bortezomib, melphalan and dexamethasone (BMDex) for untreated patients with AL amyloidosis. Blood 2016; 128 : 646.
27. Kaufman GP, Schrier SL, Lafayette LA, et al. Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis. Blood 2017; 130 : 900–902.
28. Kumar SK, Hayman SR, Buadi FK, et al. Lenalidomide, cyclophosphamide and dexamethasone (CRd) for light chain amyloidosis: long term results from a phase 2 trial. Blood 2012; 112 : 4860–4867.
29. Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: Colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336 : 1202–1207.
30. Kyle RA. Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23 : 3–9.
31. Lagos GG, Lentzsch S, Comenzo RL, et al. Final results of a phase 2 study of bendamustine in combination with dexamethasone in patients with previously treated systemic light-chain (AL) amyloidosis. Blood 2016; 128 : 4523.
32. Landau H, Hassoun H, Rosenzweig MA, et al. Bortezomib and dexamethasone consolidation following risk-adapted melphalan and stem cell transplantation for patients with newly diagnosed light-chain amyloidosis. Leukemia 2013; 27(4): 823–828.
33. Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108 : 2520–2530.
34. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologix response in patients with AL amyloidosis. Blood 2012; 119 : 4391–4394.
35. Milani P, Schönland S, Merlini G, et al. Treatment of AL amyloidosis with bendamustine: a study of 122 patients. Blood 2018; 132 : 1988–1991.
36. Moreau P. Autologous stem cell transplantation for AL amyloidosis: a standard therapy? Leukemia 1999; 13 : 1929–1931.
37. Moreau P, Jaccard A, Benboubker L, et al. Lenalidomide in combination with melphalan and dexamethasone in patiens with newly-diagnosed AL amyloidosis: a multicenter phase ½ dose escalation study. Blood 2010; 116 : 4777–4782.
38. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood 2004; 103 : 2936–2938.
39. Palladini G, Perfetti V, Perlini S, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood 2005; 105 : 2949–2951.
40. Palladini G, Russo P, Nuvolone M, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood 2007; 110 : 787–788.
41. Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood 2016; 128 : 159–168.
42. Palladini G, Russo P, Lavatelli F, et al. Treatment of patients with advanced cardiac AL amyloidosis with oral melphalan, dexamethasone and thalidomide. Ann Hematol 2009; 88 : 347–350.
43. Palladini G, Folli A, Milani P, et al. Oral melphalan and dexamethasone for AL amyloidosis: efficaccy, prognostic factors and response criteria. Amyloid 2010; 17 : 81–82.
44. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012; 30 : 4514–4549. (Palladini 2012a)
45. Palladini G, Russo P, Foli A, et al. Salvage therapy with lenalidomide and dexamethasone in patients with advanced AL amyloidosis refractory to melphalan, bortezomib, and thalidomide. Ann Hematol 2012; 91 : 89–92. (Palladini 2012b)
46. Palladini G, Russo P, Milani P, et al. A phase II trial of cyclophosphamide, lenalidomide and dexamethasone in previously treated patiens with AL amyloidosis. Haematologica 2013; 98 : 433–436.
47. Palladini G, Sachchithanantham S, Milani P, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood 2015; 126(5): 612–615.
48. Reece DE, Hegenbart U, Sanchorawala V, et al. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase ½ study. Blood 2011; 118 : 865–873.
49. Richards DB, Cookson LM, Berges AC, et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med 2015; 373(12): 1106–1114.
50. Ryšavá R, Straub J, Vacková B, et al. Results of autologous stem cell transplantation for AL amyloidosis in one Czech center. Amyloid. 2011; 18(Suppl 1): 139–141.
51. Ryšavá R. AL amyloidosis: advances in diagnostics and treatment. Nephrol Dial Transplant 2018 Oct 8. doi: 10.1093/ndt/gfy291
52. Sanchorawala V, Skinner M, Quillen K, et al. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantatrion. Blood 2007; 110 : 3561–3563. (Sanchorawala 2007a)
53. Sanchorawala V, Wright D, Rosenzweig M, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: resutls of a phase 2 trial. Blood 2007; 109 : 492–496. (Sanchorawala 2007b)
54. Sanchorawala V, Wright DG, Quillen K, et al. Tandem cycles of high-dose melphalan and autologous stem cell transplantation increase the response rate in AL amyloidosis. Bone Marrow Transplant 2007; 40(6): 557–562. (Sanchorawala 2007c)
55. Sanchorawala V, Sun F, Quillen K, et al. Long-term outcome of patients with amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 2015; 126(20): 2354–2347.
56. Sanchorawala V, Shelton AC, Lo S, Varga C, Sloan JM, Seldin DC. Pomalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 1 and 2 trial. Blood 2016; 128(8): 1059–1062.
57. Sanchorawala V, Palladini G, Kukreti V, et al. A phase ½ study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood 2017; 130(5): 597–605.
58. Seldin D, Choufani E, Dember L, et al. Tolerability and efficacy of thalidomide for the treatment of patients with light chain-associated (AL) amyloidosis. Clinical Lymphoma 2003; 3 : 241–246.
59. Sidana S, Narkhede M, Elson P, et al. Neuropathy and efficacy of once weekly subcutaneous bortezomib in multiple myeloma and light chain (AL) amyloidosis. Plos One 2017; 9; 12(3): e0172996.
60. Sidiqi MH, Aljama MA, Buadi FK, et al. Stem cell transplantation for light chain amyloidosis: decreased early mortality over time. J Clin Oncol 2018; 36 : 1323–1329.
61. Skinner M, Anderson J, Simms R, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone and colchicine versus colchicine only. Am J Med 1996; 100 : 290–298.
62. Skinner M, Sanchorawala V, Seldin D, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Int Med 2004; 140 : 85–93.
63. Specter R, Sanchorawala V, Seldin DC, et al. Kidney dysfunction during lenalidomide treatment for AL amyloidosis. Nephrol Dial Transplant 2011; 26(3): 881–886.
64. Tapan U, Seldin DC, Finn KT, et al. Increases in B-type natriuretic peptide (BNP) during treatment with lenalidomide in AL amyloidosis. Blood 2010; 116(23): 5071–5072.
65. Venner CP, Lane T, Foard D, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood 2012; 119 : 4387–4390.
66. Venner CP, Gillmore JD, Sachchithanantham S, et al. A matched comparison of cyclophosphamide, bortezomib and dexamethasone (CVD) versus risk-adapted cyclophosphamide, thalidomide and dexamethasone (CTD) in AL amyloidosis. Leukemia 2014; 28(12): 2304–2310.
67. Vesole DH, Perez WS, Akasheh M, et al. High-dose therapy and autologous hematopoietic stem cell transplantation for patients with primary systemic amyloidosis: A Center for International Blood and Marrow Transplant Research Study. Mayo Clin Proc 2006; 81 : 880–888.
68. Wechalekar AD, Goodman HJ, Lachmann HJ, et al. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood 2007; 109 : 457–464.
69. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 2013; 121 : 3420–3427.
70. Wechalekar AD, Gillmore JD, Bird J, et al. Guidelines on the management of AL amyloidosis. Brit J Hematol 2015; 168 : 186–206. (Wechalekar 2015a)
71. Wechalekar AD, Whelan C, Lachmann H, et al. Oral doxycycline improves outcomes of stage III AL amyloidosis – a matched case control study. Blood 2015; 126 : 732. (Wechalekar 2015b)
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2019 Číslo Supplementum 1
- Dentální extrakce u hemofiliků
- Sport nemění u zdravých mužů skóre HJHS
- Rizikové faktory pro rozvoj inhibitorů u dětí s hemofilií – výsledky kohortové studie
- Kazuistika: Neobvyklá příčina prodlouženého APPT u SLE – získaná hemofilie a lupus antikoagulans
- Ekonomický význam léčby krvácení u pacientů s inhibitorem pomocí rFVIIa
Najčítanejšie v tomto čísle
- DIAGNOSTIKA A LÉČBA SYSTÉMOVÉ AL AMYLOIDÓZY
- DIAGNOSTIKA A LÉČBA WALDENSTRÖMOVY MAKROGLOBULINEMIE
- ÚVODNÍ SLOVO
- ÚVODNÍ SLOVO