
Chronická myelomonocytová leukémia – prehľad súčasných diagnostických a liečebných možností
Chronic myelomonocytic leukemia – a review of present diagnostic and treatment options
Chronic myelomonocytic leukemia (CMML) is a clonal disorder of hematopoietic stem cell characterized by features of a myelodysplastic syndrome (MDS) and a myeloproliferative neoplasm (MPN). It occurs mainly in older people, mostly in men, and often transforms into acute myeloid leukemia (AML). The most common symptoms of CMML reflect hypercatabolism, the present bicytopenia, (hepato)splenomegaly and organ infiltration with monocytes. It is characterized by absolute monocytosis (> 1 x 109/l) in the peripheral blood that persists longer than 3 months. Cytogenetic examination confirmed the presence of chromosomal aberrations in approximately 40% of cases, but they are not specific for this disease. Nowadays, significant attention is focused on the TET2 gene mutation – its prognostic significance is not fully understood. JAK2 (V617F) mutation is present mainly in myeloproliferative variant of MP-CMML. The disease has variable clinical course and a very poor prognosis. Although new information from molecular biology of CMML are implemented to the treatment strategy, the results are still unsatisfactory. The objective of this review is to summarize recent information about the disease pathogenesis and the proposal for a treatment algorithm.
Key words:
chronic myelomonocytic leukemia, diagnostics, pathogenesis, TET2 gene, therapy
Autoři:
P. Rohoň; M. Jarošová; K. Indrák
Působiště autorů:
Hemato-onkologická klinika FN a LF UP v Olomouci
Vyšlo v časopise:
Transfuze Hematol. dnes,20, 2014, No. 2, p. 58-66.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Chronická myelomonocytová leukémia (CMML) patrí do skupiny klonových ochorení hematopoetickej kmeňovej bunky a nesie črty myelodysplastického syndrómu (MDS) i myeloproliferatívnych neoplázii (MPN). Vyskytuje sa predovšetkým u starších osôb, prevažne u mužov, a v mnohých prípadoch transformuje do akútnej myeloblastovej leukémie (AML). Najčastejšie klinické príznaky CMML odrážajú hyperkatabolizmus, prítomnú bicytopéniu, (hepato)splenomegáliu a orgánovú infiltráciu monocytmi. V periférnej krvi je charakteristická absolútna monocytóza (> 1 x 109/l), ktorá pretrváva dlhšie ako 3 mesiace. Cytogenetické vyšetrenie potvrdzuje prítomnosť chromozómových aberácii až v 40 % prípadov, ktoré ale nie sú pre toto ochorenie špecifické. V poslednom období je venovaná veľká pozornosť mutácii TET2 génu – jej prognostický význam však nie je úplne objasnený. JAK2 (V617F) mutácia je prítomná najmä u myeloproliferatívnej varianty CMML (MP-CMML). Ochorenie má variabilný klinický obraz a veľmi zlú prognózu. Hoci existuje snaha o implementáciu poznatkov z molekulovej biológie CMML do stratégie liečby, jej výsledky sú stále neuspokojivé. Cieľom prehľadového článku je zhrnúť najnovšie poznatky o patogenéze ochorenia a vytvoriť návrh liečebného algoritmu.
Klíčová slova:
chronická myelomonocytová leukémia, diagnostika, patogenéza, TET2 gén, liečba
Úvod
CMML je ochorenie, ktoré sa vyskytuje s incidenciou 1,5–3,5/100 000 obyvateľov a rok. Je časté u starších osôb s vekovým mediánom medzi 65–75 rokom, častejšie u mužov (muži : ženy – 2 : 1), s pravdepodobnosťou transformácie do AML 20 % a s mediánom celkového prežitia (OS) neliečených pacientov nepresahujúcim 20 mesiacov. Je to ochorenie hematopoetickej kmeňovej bunky s črtami myelodysplázie i myeloproliferácie charakterizované predovšetkým perzistujúcou monocytózou v periférnej krvi (> 1 x 109/l), neprítomnosťou chromozómu Philadelphia alebo fúzneho génu BCR/ABL1, bez prestavby génov PDGFR-a, PDGFR-b a počtom blastov v kostnej dreni (KD) nepresahujúcim 20 % (1). Asi v 80 % vzniká CMML de novo, v ostatných prípadoch je v anamnéze MDS, často i s monocytózou. Ochorenie má variabilný priebeh i klinický obraz – najčastejšie príznaky sú dôsledkom prítomnej cytopénie, (hepato)splenomegálie (30–50 % prípadov), orgánovej infiltrácie monocytmi a nádorovým katabolizmom (2). Asi v 40 % prípadov CMML sú prítomné cytogenetické aberácie, ktoré však nie sú pre toto ochorenie špecifické. Mezi častými genetickými zmenami sa vyskytujú uniparentálne dizómie (UPD) (50 %), ktoré môžu byť združené s mutáciami CBL génu (3), poruchou zostrihového mechanizmu (spliceozóm) – mutácie SRSF2 génu a napokon s mutáciami ASXL1 a RUNX (4). Je však veľmi zložité rozhodnúť, ktoré zo somatických mutácii sú priamo zodpovedné za patogenézu ochorenia a ktoré sú dôsledkom progresie CMML. V poslednom období sa do popredia dostáva význam mutácie génu TET2 (Ten-Eleven Translocation-2) a predovšetkým jej súvislosť s liečbou hypometylačnými látkami. Prítomnosť mutácie je popisovaná u myeloidných neoplázii zahŕňajúc myeloproliferácie i MDS (5, 6). V patogenéze CMML sa uplatňuje akumulácia génových mutácii, v hematopoetických prekurzoroch s najvyšším počtom mutácii dochádza k alterovanej granulocytovej a monocytovej diferenciácii. Najvyšší počet genetických abnormalít sa zistil u myeloproliferatívnej formy CMML (napr. TET2, IDH, SRSF2, ASXL1) (7). Výsledky liečby CMML sú neuspokojivé a súvisia s biologickou podstatou choroby a obmedzeným repertoárom cielenej terapie. V zásade jedinou možnosťou predĺženia prežitia a príp. i vyliečenia je alogénna transplantácia hematopoetických kmeňových buniek (ASCT), je však dostupná iba pre obmedzenú skupinu osôb vzhľadom na vysoký vek pri diagnóze a časté komorbidity. Nemenej dôležitým faktom je aj vysoký počet relapsov po ASCT (8, 9). Medzi základné, predovšetkým cytoredukčné lieky, patrí hydroxyurea (HU), etopozid a nízkodávkovaný cytozín-arabinozid (Ara-C) (10). V podpornej liečbe sa využívajú erytropoézu stimulujúce proteíny (ESP) a granulocytové rastové faktory (G-CSF). Veľké očakávania sú vkladané do použitia hypometylačných látok (5-azacytidín, 5-AZA a decitabín, DAC) a ich kombinácie s novými molekulami, doposiaľ však chýbajú výsledky väčších randomizovaných štúdií (11). V ojedinelých prípadoch potvrdenej prestavby génu PDGFR-b bol potvrdený účinok imatinibu – podľa poslednej WHO klasifikácie však tieto prípady nie sú klasifikované ako CMML (12).
Diagnostika a klasifikácia
CMML má veľmi variabilný klinický priebeh (13, 14). Ochorenie môže byť odhalené náhodne pri vyšetrení krvného obrazu z inej príčiny, častejšie sa však objavujú príznaky vyplývajúce z prítomnej (bi)cytopénie, splenomegálie či konštitučné symptómy. Lokalizované alebo generalizované zväčšenie lymfatických uzlín je u CMML zriedkavé, vyskytuje sa častejšie u detí (JMML), vo všeobecnosti koreluje s progresiou ochorenia (15). V zásade však prvotným ukazovateľom zostáva dlhodobo pretrvávajúca periférna monocytóza (> 1 x 109/l) (po vylúčení infekčných a autoimunitných príčin), ktorá vedie k pokračovaniu diagnostického algoritmu hematológom. Základným nástrojom pre upresnenie diagnózy je aspirát KD s nálezom myelomonocytov, ktoré nemajú morfológiu klasických monocytov, ale nesú i rysy granulocytového radu (príbuznosť CMML a MDS), dysplázie v ostatných hematopoetických radoch (obr. 1) a/alebo prítomnosťou klonových genetických aberácii. K pomocným vyšetreniam patrí prietoková cytometria aspirátu a histologické vyšetrenie trepanobioptickej vzorky. Komplementárny význam imunohistochémie a prietokovej cytometrie tkvie predovšetkým v kvantifikácii počtu blastov a taktiež v popise aberantnej expresie znakov monocytovej línie. Detailná laboratórna analýza má mimoriadny význam pre stanovenie prognózy pacienta.

Klasifikácia CMML prekonala viacero vývojových etáp. Diagnostické kritériá FAB zaradili CMML do skupiny MDS a pre potvrdenie diagnózy boli stanovené nasledujúce kritériá: periférna monocytóza (> 1 x 109/l), počet blastov v KD < 20 %, prítomnosť uni-/bi-/trilineárnej dysplázie, počet periférnych blastov < 5 % a neprítomnosť Auerových tyčí v myeloidnej línii (16). Ďalším spresnením FAB klasifikácie bolo rozdelenie CMML na dve varianty: myelodysplastickú (MD-CMML) a myeloproliferatívnu (MP-CMML) so zohľadnením počtu leukocytov a stanovením hranice (13 x 109/l). Toto rozdelenie malo predovšetkým prognostický význam pre OS, avšak nestratifikovalo pacientov s ohľadom na možnosť transformácie do AML (17, 18). Na tomto mieste je dôležité pripomenúť, že rozdelenie na spomínané varianty má významný vplyv na výber liečby – predovšetkým epigenetickej (hypometylačnej). V súčasnosti je podľa WHO klasifikácie 2008 CMML zaradená do skupiny myelodysplastických/myeloproliferatívnych neoplázii a obsahuje 2 podskupiny: CMML-1 (< 5 % blastov i promonocytov v periférii a < 10 % blastov v KD) a CMML-2 (5–19 % blastov v periférii a 10–19 % blastov v KD alebo prítomnosť Auerových tyčí) (19) (tab. 1). Dôležité je tiež poukázať na skutočnosť, že časť MDS pacientov má prítomnú monocytózu, čo je jedným z dôvodov blízkosti MDS a CMML. Nie je vzácnosťou, že niektorí pacienti, ktorí prichádzajú ako MDS nízkeho rizika s cytopéniou a monocytózou prechádzajú počas rôzne dlhého časového intervalu do obrazu MP-CMML (18).

Cytogenetické vyšetrenie potvrdzuje prítomnosť chromozómových aberácii asi v 40 % prípadov, ktoré však nie sú pre toto ochorenie špecifické. Najčastejšie sú pozorované abnormality chromozómu 7 (monozómie), 8 (trizómie) a komplexné prestavby (20, 21). Molekulovo-genetické zmeny je možné rozčleniť do niekoľkých podskupín: epigenetická regulácia (TET2, DNMT3A), transkripcia (RUNX1), poruchy zostrihového mechanizmu (SRSF2), signálne dráhy tyrozínových kináz (NRAS, KRAS, CBL, JAK2) a ďalšie (napr. ASXL1) (22). Medzi najčastejšie mutácie u CMML pacientov patria: TET2 (58 %), SRSF2 (47 %) a ASXL1 (38 %) (4).
Variabilita CMML nie je podmienená iba širokým spektrom cytogenetických a molekulovo-genetických zmien, ale tiež skutočnosťou, že do obrazu CMML sú na jednej strane zahrnuté klinicky nezávažné cytopénie s monocytózou a na strane druhej vzácnejšie ťažké formy s nádorovým rastom myelomonocytových infiltrátov v koži, pľúcach a iných orgánoch. Klasický IPSS systém používaný pre pacientov s MDS (23) nezohľadňuje niektoré dôležité charakteristiky CMML – predovšetkým v prípade MP-CMML – ale tiež vek pacienta, performance status, transfúznu závislosť. Preto bol navrhnutý celý rad skórovacích systémov, poslednou variantou je CPSS (CMML-specific prognostic scoring system), ktorý rozdeľuje pacientov do 4 prognostických skupín a obsahuje napr. cytogenetickú klasifikáciu rizikových podskupín, transfúznu závislosť, WHO CMML podtypy (24, 25) (tab. 2). V poslednom období nastal veľký rozmach informácii o prognostickom význame niektorých somatických mutácii či zmien ich expresie: napr. mutácia ASXL1 – rýchlejšia evolúcia do AML (26), znížená expresia CJUN – zlepšenie OS pri liečbe decitabínom (DAC) (27). Zatiaľ však vo všeobecnosti nie sú súčasťou prognostických skórovacích systémov, pretože ich prítomnosť je kontroverzná (napr. TET2).

Patogenéza
V súčasnosti je pohľad na patogenézu CMML ovplyvnený identifikáciou širokej palety molekulových zmien a informáciami zo zvieracieho modelu MDS (28). CMML je ochorením hematopoetickej kmeňovej bunky pričom dochádza k lineárnej akumulácii génových mutácii – prvotný zásah pravdepodobne postihuje gén TET2 (iniciálna klonová dominancia zasahujúca epigenetické mechanizmy, ktoré vyústia do predčasnej granulocytovej/monocytovej (GM) diferenciácie) (7). GM hyperplázia vzniká ako dôsledok ďalších mutácii v CBL/RAS signálnej dráhe (29). Akumulácia mutácií vedie často ku koexistencii jednotlivých podskupín: onkogénne mutácie (napr. RUNX1) sa vyskytujú súčasne s mutáciami zasahujúcimi epigenetické regulácie (napr. TET2, ASXL1). V konečnom dôsledku dochádza ku kumulácii maturovaných CD14+ monocytov a dysplastických granulocytov. Otvorenou otázkou zostáva mechanizmus prechodu do AML (obr. 2).

V prípade MDS, CMML a JMML je častým fenoménom výskyt spoločných mutácii, ktoré môžu mať čiastočne odlišný fenotypový prejav (30, 31). Po určitom zjednodušení je možné vyčleniť u pacientov s CMML v zásade dve skupiny zmien: somatické mutácie génov (tab. 3) a rekurentné cytogenetické abnormality (balansované alebo nebalansované). Balansované translokácie sú zriedkavejšie a zahŕňajú prestavby PDGFRB génu (5q31-33) s vytvorením fúzie ETV6/PDFGRB. Podľa WHO klasifikácie 2008 patria jednotky s prestavbami génov PDGFRA, PDGFRB a FGFR1 (vylučovacie kritérium CMML) do skupiny myeloidných neoplázii s eozinofíliou, nadôvažok pacienti s PDGFRB prestavbou sú kandidátmi liečby pomocou inhibítorov TK (12). Nebalansované abnormality sa s využitím konvenčnej cytogenetiky vyskytujú až v 40 % prípadov CMML, nie sú pre toto ochorenie špecifické a čiastočne sa prekrývajú so zmenami u MDS. Je to najmä trizómia 8, monozómia 7 či delécia 7q a delécia 20q (30). Pozorované boli tiež komplexné karyotypy, delécia 12p (38) alebo hypodiploidný karyotyp (37–39). Such a kol. potvrdili prognostický význam chromozómových zmien u CMML (40). Analýza obsahovala rozdelenie pacientov podľa prítomných zmien na skupinu „low risk“ (normálny karyotyp a strata Y ako jediná zmena), „high risk“ (trizómia 8, zmeny chromozómu 7 alebo komplexný karyotyp) a napokon „intermediate risk“ (všetky ďalšie zmeny). Klinické potvrdenie tejto stratifikácie sa stále očakáva. Využitím sofistikovaných genetických metód (napr. SNP array) je u CMML tiež možné identifikovať stratu heterozygozity v dôsledku získanej UPD a kryptických chromozómových abnormalít (41).

Liečba
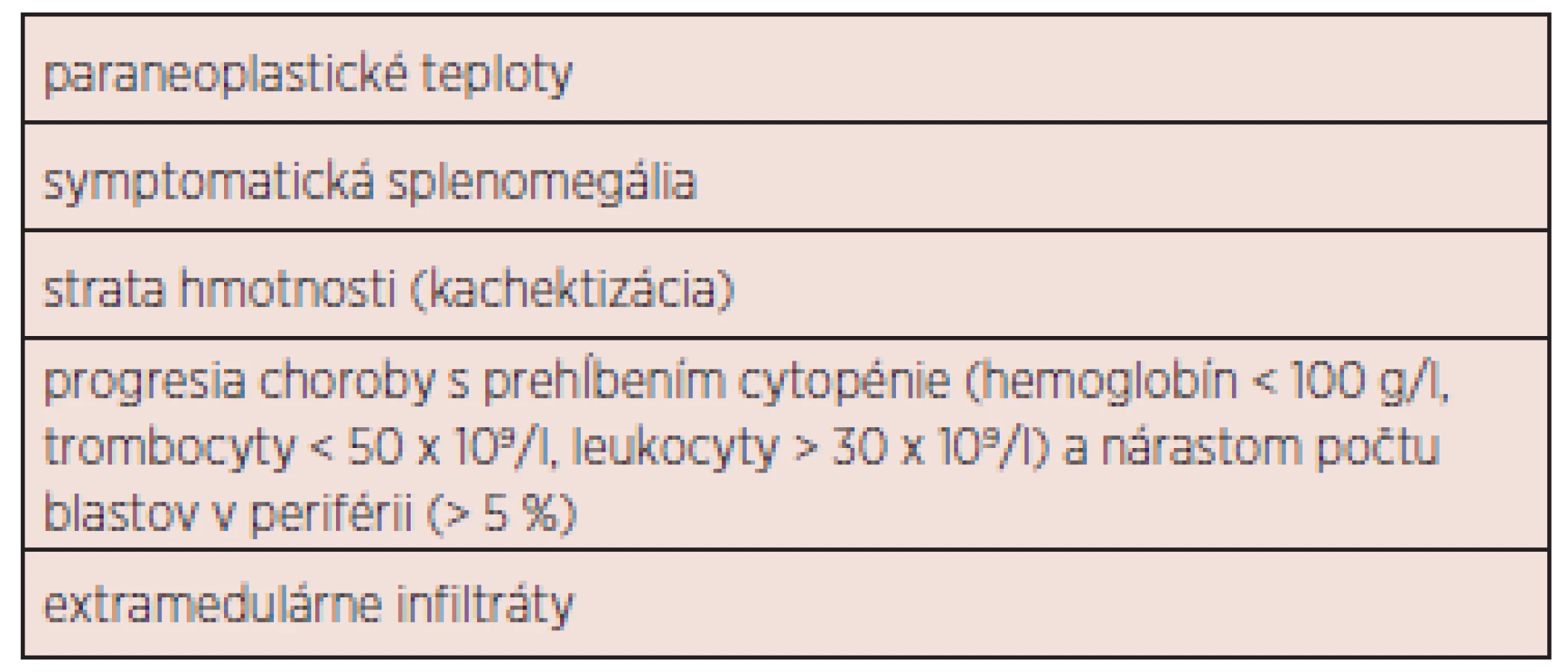
Výsledky liečby CMML sú neuspokojivé. Doposiaľ neboli vytvorené univerzálne terapeutické odporúčania. Základnou otázkou je správne načasovanie zahájenia liečby (tab. 4). V súčasnosti môžeme liečebné možnosti rozdeliť do 4 základných podskupín: 1. observácia (asymptomatická monocytóza, do stabilizácie počtu leu-kocytov kontrola KO jedenkrát mesačne, neskôr v trojmesačnom intervale), 2. podporná liečba, 3. cytoredukcia, chemoterapia, hypometylačná (epigenetická) liečba 4. ASCT. Liečebný plán by mal byť jasne definovaný, najmä v prípade plánovanej ASCT. Nemenej významnou otázkou je spôsob hodnotenia liečebnej odpovede. Onida a kol. odporúčajú využiť pre MD-CMML kritériá IWG pre MDS 2006 (42), pre MP-CMML kritériá IWG pre primárnu myelofibrózu 2009 (43). Rezistencia k liečbe pomocou 5-AZA pri MD-CMML by mala byť hodnotená po 6 cykloch (21). Súhrnný návrh prehľadu liečby 1. a 2. línie ukazuje obrázok 3. Žiaľ, terapia pacientov s CMML zostáva vo väčšine prípadov i s ohľadom na vek pacientov podporná a cytoredukčná. V ďalšom texte budú diskutované jednotlivé liečebné modality samostatne.

Alogénna transplantácia hematopoetických kmeňových buniek
ASCT je jedinou kuratívnou metódou v liečbu CMML. Jej realizácia je však limitovaná vysokým vekom a komorbiditami pacientov. Všeobecne je ASCT u CMML spojená s vysokou úmrtnosťou, TRM (25–40 %). ASCT s režimami s redukovanou intenzitou (RIC) nemá u týchto pacientov vyšší výskyt relapsov v porovnaní s klasickým prípravným režimom (44, 45), zlepšuje OS a umožňuje transplantovať i starších pacientov. OS po 5 rokoch po transplantácii osciloval medzi 18–75 % – podľa typu štúdie (21). Retrospektívna EBMT analýza potvrdila dôležitosť dosiahnutia kompletnej remisie pred ASCT – zníženie výskytu potransplantačných relapsov (9). V prípade MP-CMML je vhodné zahájiť cytoredukciu blastov použitím polychemoterapeutického režimu („AML like“), v prípade MDS-CMML je možné zvážiť premostenie pomocou 5-AZA.
Hydroxyurea
HU je 1. líniou liečby starších pacientov s nízkym počtom blastov (< 10 %) v KD. Cieľom tejto liečby je redukovať príznaky choroby, avšak bez ambície predĺžiť OS. Pri porovnaní HU (1 g/deň) a etopozidu (150 mg/týždeň) v súbore 105 pacientov bol pozorovaný vyšší počet liečebných odpovedí (60 % vs. 30 % po 11 mesiacoch sledovania) a OS (20 vs. 9 mesiacov) (10). U pacientov, ktorí sú rezistentní k podaniu HU, je vhodné v 2. línii zvážiť LD-AraC. Podanie 5-AZA je spojené s vyšším výskytom nežiaducich účinkov (21).
Hypometylačné látky
V súčasnosti sa pre liečbu CMML používajú 2 hypometylačné látky: 5-azacytidín a decitabín. 5-AZA je liekom 1. voľby u pacientov s CMML s počtom blastov v KD 10–29 % a bez známok myeloproliferácie. Odporúča sa podať minimálne 6 cyklov liečby (súhlasné odporučenie FDA i EMEA). Pilotnú štúdiu s využitím 5-AZA publikoval Silverman et al., bolo do nej zaradených 361 pacientov s MDS (CMML malo iba 14 z nich) a hoci výsledky CMML pacientov neboli uvádzané zvlášť, celková liečebná odpoveď (ORR) nemala u podskupín RA, RARS, REAB i CMML štatisticky signifikantný rozdiel (46). V súčasnosti sú už dostupné výsledky väčšieho počtu klinických štúdii (retrospektívnych), ktoré dokumentujú predovšetkým OS (osciluje od 1–3 rokov), ORR (25–70 %) a toxický profil (47) (tab. 5). Dôležitým momentom pri podávaní hypometylačních látok je identifikácia prediktora klinickej odpovede. Existujú racionálne podklady pre podanie 5-AZA u pacientov s mutáciou TET2 (bez súčasného výskytu mutácie ASXL1), hoci neboli štatisticky potvrdené. Pomocou multivariačnej analýzy boli ako negatívne prediktory liečebnej od-povede označené tiež blasty v KD > 10 % a počet leukocytov > 13 x 109/l. Použitie 5-AZA u MP-CMML je stále predmetom diskusie, najmä preto, že pri nízkom dávkovaní má predovšetkým demetylační účinok s minimálnou toxicitou a nedokáže ovplyvňovať zvýšenú proliferačnú aktivitu (21). DAC spôsobuje pri nízkom dávkovaní hypometyláciu určitých génov (napr. p15INK4b, HIC1, p21CIP1, p57KIP2, čo je dôležité pre reaktiváciu tumorsupresorových génov) (53). V niektorých krajinách EU je využívaný pre liečbu AML u starších pacientov alebo MDS vyššieho rizika bez možnosti ASCT pri zlyhaní iných modalít vrátane 5-AZA. Podáva sa formou i.v. infúzie (obvykle 15 mg/m2 počas 3 dní), jeho odpoveď sa hodnotí po 4 cykloch. Prvé štúdie s DAC publikoval Kantarjian a kol., v malom súbore 7 pacientov s CMML bola zaznamenaná odpoveď iba u jedného (54). Štúdie s využitím DAC vykazujú ORR medzi 40–70 % (11, 55). Zdá sa, že prediktorom liečebnej odpovede by mohla byť nízká expresia CJUN a CMYB (56). Hypometylačné látky majú prijateľný toxický profil a ich kombinácia s novými molekulami (napr. inhibítory HIDAC) môže predstavovať prielom v netransplantačnej liečbe CMML. Otvorenými otázkami však naďalej zostávajú: dávkovacia schéma, terapia po dosiahnutí CR a využitie 5-AZA v predtransplantačnom režime a liečbe potransplantačného relapsu.

Intenzívna chemoterapia
V prípade CMML existuje najväčšie množstvo informácii o podaní stredných dávok Ara-C s topotekanom. Táto kombinácie bola podávaná pacientom s MP-CMML s vyšším počtom blastov. CR bola dosiahnutá asi u 40 % pacientov, medián OS bol 10 mesiacov (57). V prípade sólo terapie topotekanom bola CR dosiahnutá v 28 % a medián OS bol 10 mesiacov (58).
Ďalšie liečebné možnosti
Podanie kortikoidov nie je možné všeobecne odporučiť, sporadicky bol popísaný ich účinok u pacientov s trombocytopéniou a reumatoidnými príznakmi. Thalidomid má pluripotentný účinok (inhibícia TNF-α, antiangiogénny efekt) a u MDS nízkeho rizika bola popísaná erytroidná odpoveď, u vyššieho rizika prechod do AML. Klofarabín je purinovým analógom, inhibuje syntézu DNA a ribonukleoitidovej reduktázy. Jeho účinok bol potvrdený u MDS vyššieho rizika (59). V prípade mutácii N-RAS a K-RAS (predovšetkým MP-CMML) existuje racionálny podklad pre podávanie inhibítorov farnezylovej transferázy (FT) (60). Strupp a kol. podali v pilotnom projekte bendamustín 15 starším predliečeným pacientom s AML a MDS vyššieho rizika pričom bol dokázaný jeho cytoredukčný potenciál, nebolo však potvrdené dosiahnutie hematologickej odpovede či zníženie transfúznej závislosti (61).
Diskusia a záver
CMML je zriedkavé ochorenie prevažne starších osôb s doposiaľ veľmi nejasnou etiopatogenézou vzniku a veľmi zlými výsledkami liečby. V poslednom období sa však s využitím nových technológii podarilo zistiť, že značná časť pacientov (predovšetkým MP-CMML) nesie mutácie v epigenetických regulačných génoch (TET2, ASXL1, EZH2), ktoré modifikujú kondenzáciu chromatínu a v konečnom dôsledku i génovú expresiu. Rôznorodá kombinácia mutácii podporuje teóriu viacstupňovej (multistep) patogenézy a variabilného klinického priebehu (31). Hoci je pravdou, že tieto poznatky zatiaľ nie sú zahrnuté do klinických aplikácii, objavujú sa prvé práce, ktoré tieto informácie implementujú do skórovacích systémov (napr. gén ASXL1) (62). Pre jasné stanovenie významu jednotlivých zmien bude, pochopiteľne, nutné vyšetriť väčšie kohorty pacientov. V liečbe CMML je zjavné najvýznamnejšie postavenie ASCT ako jedinej kuratívnej modality, so zavedením RIC sa jej použitie posúva do vyšších vekových kategórií (44, 45). Žiaľ, ASCT i tak zostává minoritnou možnosťou pre väčšinu pacientov a preto sa upierajú nové nádeje k využitiu hypometylačných látok. Ich aplikácia zohladňuje niekoľko recentných odporučení (1, 14, 21). Súhrnom je možné konštatovať, že základnou výzvou pre zlepšenie celkového prežívania pacientov do budúcnosti je preklenúť priepasť medzi narastajúcimi informáciami o molekulovej patogenéze choroby a možnosťami cielenej liečby.
Zoznam použitých skratiek
- 5-AZA – 5-azacytidín
- ASCT – alogénna transplantácia hematopoetických kmeňových buniek
- C – cytozín
- CMML – chronická myelomonocytová leukémia
- CR – kompletná remisia
- DAC – decitabín
- EPO/ESP – erytropoézu stimulujúce proteíny
- FT – farnezylová transferáza
- G-CSF – granulocytový rastový faktor
- GM – granulocytový/monocytový
- HI – hematologické zlepšenie
- HU – hydroxyurea
- KD – kostná dreň
- LD-AraC – nízkodávkovaný cytozínarabinozid
- MD-CMML – myelodysplastická varianta CMML
- MDS – myelodysplastický syndróm
- MP-CMML – myeloproliferatívna varianta CMML
- MPN – myeloproliferatívne neoplázie
- OS – celkové prežitie
- PK – periférna krv
- RIC – režim s redukovanou intenzitou
- TK – tyrozínová kináza
- TRM – mortalita spojená s transplantáciou
- UPD – uniparentálna dizómia
Práca bola podporená grantom IGA UP LF-2014-001 a NT12218-4.
P. Rohoň – príprava rukopisu
M. Jarošová – revízia rukopisu
K. Indrák – revízia rukopisu
Doručeno do redakce: 19. ledna 2014
Přijato po recenzi: 7. května 2014
MUDr. Peter Rohoň, Ph.D.
Hemato-onkologická klinika FN a LF UP v Olomouci
I. P. Pavlova 6
775 20 Olomouc
e-mail: peter.rohon@centrum.cz
Zdroje
1. Parikh SA, Tefferi A. Chronic myelomonocytic leukemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87 : 610-619.
2. Tefferi A, Elliott MA, Pardanani A. Atypical myeloproliferative disorders: diagnosis and management. Mayo Clin Proc 2006; 81 : 553-563.
3. Dunbar AJ, Gondek LP, O’Keefe CL, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 2008; 68 : 10349-10357.
4. Kosmider O, Gelsi-Boyer V, Ciudad M, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009; 94 : 1676-1681.
5. Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009; 114 : 144-147.
6. Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009; 23 : 1343-1345.
7. Perez C, Martinez-Calle N, Martin-Subero JI, et al. TET2 mutations are associated with specific “5“-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS ONE 2012; 7: e31605.
8. Kroger N, Zabelina T, Guardiola P, et al. Allogeneic stem cell transplantation of adult chronic myelomonocytic leukaemia. A report on behalf of the Chronic Leukaemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Br J Haematol 2002; 118 : 67-73.
9. Ocheni S, Kroger N, Zabelina T, et al. Outcome of allo-SCT for chronic myelomonocytic leukemia. Bone Marrow Transplant 2009; 43 : 659-661.
10. Wattel E, Guerci A, Hecquet B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Francais des Myelodysplasies and European CMML Group. Blood 1996; 88 : 2480-2487.
11. Aribi A, Borthakur G, Ravandi F, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer 2007; 109 : 713-717.
12. Pitini V, Arrigo C, Teti D, et al. Response to STI571 in chronic myelomonocytic leukemia with platelet derived growth factor beta receptor involvement: a new case report. Haematologica. 2003; 88: e78-e79.
13. Germing U, Kundgen A, Gattermann N. Risk assessment in chronic myelomonocytic leukemia (CMML). Leuk Lymphoma 2004; 45 : 1311-1318.
14. Onida F, Kantarjian HM, Smith TL, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood 2002; 99 : 840-849.
15. Bizet M, Callat MP, Goasguen J, et al. Chronic myelomonocytic leukemia with lymphadenopathy. Myelodysplastic Syndromes, Springer, 1992.
16. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982; 51 : 189-199.
17. Bennett JM, Catovsky D, Daniel MT, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol 1994; 87 : 746-754.
18. Voglová J, Chrobák L, Neuwirtová R, et al. Myelodysplastic and myeloproliferative type of chronic myelomonocytic leukemia - distinct subgroups or two stages of the same disease? Leuk Res 2001; 25 : 493-499.
19. Swerdlow SH, Campo E, Harris NL. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Lyon: IARC Press, 2008.
20. Bacher U, Haferlach T, Kern W, et al. Conventional cytogenetics of myeloproliferative diseases other than CML contribute valid information. Ann Hematol 2005; 84 : 250-257.
21. Onida F, Barosi G, Leone G, et al. Management recommendations for chronic myelomonocytic leukemia: consensus statements from the SIE, SIES, GITMO groups. Haematologica 2013; 98 : 1344-1352.
22. Ricci C, Fermo E, Corti S, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res 2010; 18 : 2246-2256.
23. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89 : 2079-2088.
24. Beran M, Wen S, Shen Y, et al. Prognostic factors and risk assessment in chronic myelomonocytic leukemia: validation study of the M.D. Anderson Prognostic Scoring System. Leuk Lymphoma 2007; 48 : 1150-1160.
25. Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood 2013; 121 : 3005-3015.
26. Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145 : 788-800.
27. Braun T, Itzykson R, Renneville A, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood 2011; 118 : 3824-3831.
28. Beurlet S, Chomienne C, Padua RA. Engineering mouse models with myelodysplastic syndrome human candidate genes; how relevant are they? Haematologica 2013; 98 : 10-22.
29. Kohlmann A, Grossmann V, Klein HU, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010; 24 : 3858-3865.
30. Muramatsu H, Makishima H, Jankowska AM, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 2010; 115 : 1969-1975.
31. Muramatsu H, Makishima H, Maciejewski JP. CMML and aCML: novel pathogenetic lesions. Semin Oncol 2012; 39 : 67-73.
32. Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466 : 1129-1133.
33. Makishima H, Cazzolli H, Szpurka H, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 2009; 36 : 6109-6116.
34. Kuo MC, Liang DC, Huang CF, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009; 23 : 1426-1431.
35. Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood 2012; 15: 3080-3088.
36. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365 : 1054-1061.
37. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114 : 937-951.
38. Groupe Français de Cytogénétique Hématologique. Chronic myelomonocytic leukemia: single entity or heterogeneous disorder? A prospective multicenter study of 100 patients. Cancer Genet Cytogenet 1991; 1 : 57-65.
39. Catalano L, Improta S, de Laurentiis M, et al. Prognosis of chronic myelomonocytic leukemia. Haematologica 1996; 4 : 324-329.
40. Such E, Cervera J, Costa D, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011; 96 : 375-383.
41. Gondek LP, Dunbar AJ, Szpurka H, et al. SNP array karyotyping allows for the detection of uniparental disomy and cryptic chromosomal abnormalities in MDS/MPD-U and MPD. PLoS ONE 2007; 11: e1225.
42. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108 : 419-425.
43. Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT). Blood 2006; 108 : 1497-1503.
44. Elliott MA, Tefferi A, Hogan WJ, et al. Allogeneic stem cell transplantation and donor lymphocyte infusions for chronic myelomonocytic leukemia. Bone Marrow Transplant 2006; 11 : 1003-1008.
45. Laport GG, Sandmaier BM, Storer BE, et al. Reduced intensity conditioning followed by allogeneic hematopoietic cell transplantation for adult patients with myelodysplastic syndrome and myeloproliferative disorders. Biol Blood Marrow Transplant 2008; 14 : 246-255.
46. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J Clin Oncol 2002; 20 : 2429-2440.
47. Jonášová A, Čermák J, Červinek L, et al. První zkušenosti České MDS skupiny s terapií 5-azacytidinem u nemocných s myelodysplastickým syndromem s vyšším rizikem (IPSS střední 2 a vysoké riziko), akutní myeloidní leukemií do 30 % myeloblastů a chronickou myelo-monocytární leukemií II. Transfuze Hematol dnes 2013; 3 : 125-133.
48. Costa R, Abdulhaq H, Haq B, et al. Activity of azacitidine in chronic myelomonocytic leukemia. Cancer 2011; 117 : 2690-2696.
49. Thorpe M, Montalvao A, Pierdomenico F, et al. Treatment of chronic myelomonocytic leukemia with 5-Azacitidine: A case series and literature review. Leuk Res 2012; 36 : 1071-1073.
50. Wong E, Seymour JF, Kenealy M, et al. Treatment of chronic myelo-monocytic leukemia with azacitidine. Leuk Lymphoma 2013; 54 : 878-880.
51. Fianchi L, Criscuolo M, Breccia M, et al. High rate of remissions in chronic myelomonocytic leukemia treated with 5-azacytidine: Results of an Italian retrospective study. Leuk Lymphoma 2013; 54 : 658-661.
52. Ades L, Sekeres MA, Wolfromm A, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res 2013; 37 : 609-613.
53. Plimack ER, Kantarjian HM, Issa JP. Decitabine and its role in the treatment of hematopoietic malignancies. Leukemia & Lymphoma 2007, 48 : 1472-1481.
54. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006; 106 : 1794-1803.
55. Wijermans PW, Rüter B, Baer MR, et al. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res 2008; 32 : 587-591.
56. Braun T, Itzykson R, Renneville A, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011; 118 : 3824–3831.
57. Beran M, Estey E, O’Brien S, et al. Topotecan and cytarabine is an active combination regimen in myelodysplastic syndromes and chronic myelomonocytic leukemia. J Clin Oncol 1999; 17 : 2819-2830.
58. Grinblatt DL, Yu D, Hars V, Vardiman JW, et al. Treatment of myelodysplastic syndrome with 2 schedules and doses of oral topotecan: a randomized phase 2 trial by the Cancer and Leukemia Group B (CALGB 19803). Cancer 2009; 115 : 84-93.
59. Guidelines for the diagnosis and treatment of myelodysplastic syndromes and chronic myelomonocytic leukemia. Nordic MDS Group. Issue 6. 5th update, 1st of December 2013.
60. Fenaux P, Raza A, Mufti GJ, et al. A multicenter phase 2 study of the farnesyltransferase inhibitor tipifarnib in intermediate to high-risk myelodysplastic syndrome. Blood 2007; 109 : 4158-4163.
61. Strupp C, Knipp S, Hartmann J, et al. A pilot study of bendamustine in elderly patients with high-risk MDS and AML. Leuk Lymphoma 2007; 48 : 1161-1166.
62. Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013; 31 : 2428-2436.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2014 Číslo 2
- Dentální extrakce u hemofiliků
- Sport nemění u zdravých mužů skóre HJHS
- Rizikové faktory pro rozvoj inhibitorů u dětí s hemofilií – výsledky kohortové studie
- Kazuistika: Neobvyklá příčina prodlouženého APPT u SLE – získaná hemofilie a lupus antikoagulans
- Ekonomický význam léčby krvácení u pacientů s inhibitorem pomocí rFVIIa
Najčítanejšie v tomto čísle
- Chronická myelomonocytová leukémia – prehľad súčasných diagnostických a liečebných možností
- Eliminácia rizika prenosu vírusu hepatitídy B transfúziou vo fáze možnej okultnej HBV infekcie
- Hodnocení exprese antigenu CD45 u pacientů s mnohočetným myelomem a jeho vliv na dobu do první progrese nebo relapsu
- Mechanismy navozené imunotolerance nádorových buněk u chronické lymfatické leukemie a možnosti jejich detekce metodami průtokové cytometrie