
Preferential Use of Central Metabolism Reveals a Nutritional Basis for Polymicrobial Infection
The human urinary tract is a leading source for polymicrobial infections and for the development of bacteremia and sepsis. Treating these potentially dangerous infections have recently become more challenging due to the appearance of uropathogenic strains that are resistant to the many of the most commonly prescribed antibiotics. The majority of urinary tract infections (UTI) are caused by Escherichia coli, while another bacterium, Proteus mirabilis, is more likely to cause catheter-associated UTI. Here, we report that uropathogenic E. coli and P. mirabilis have divergent nutritional requirements despite growing in the same host environment. This result indicates that E. coli and P. mirabilis do not directly compete for nutrients during UTI. Indeed, we found that persistence of both pathogens is enhanced when they co-colonize the host. This work represents an important step toward understanding the basic nutritional requirements for two major pathogens that cause UTI and shows how mixed infections can change these requirements. Understanding how bacteria grow during infections is fundamental to ultimately uncover new ways to combat increasingly drug-resistant bacterial infections.
Published in the journal:
Preferential Use of Central Metabolism Reveals a Nutritional Basis for Polymicrobial Infection. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004601
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004601
Summary
The human urinary tract is a leading source for polymicrobial infections and for the development of bacteremia and sepsis. Treating these potentially dangerous infections have recently become more challenging due to the appearance of uropathogenic strains that are resistant to the many of the most commonly prescribed antibiotics. The majority of urinary tract infections (UTI) are caused by Escherichia coli, while another bacterium, Proteus mirabilis, is more likely to cause catheter-associated UTI. Here, we report that uropathogenic E. coli and P. mirabilis have divergent nutritional requirements despite growing in the same host environment. This result indicates that E. coli and P. mirabilis do not directly compete for nutrients during UTI. Indeed, we found that persistence of both pathogens is enhanced when they co-colonize the host. This work represents an important step toward understanding the basic nutritional requirements for two major pathogens that cause UTI and shows how mixed infections can change these requirements. Understanding how bacteria grow during infections is fundamental to ultimately uncover new ways to combat increasingly drug-resistant bacterial infections.
Introduction
The recent revival of interest in the relationship between bacterial metabolism and host-pathogen interactions has deepened our understanding of pathogen colonization and growth in vivo [1], [2], [3], [4]. Consequently, central metabolism must be considered essential to virulence because bacterial pathogens must use nutrients available within the host niche to cause disease [5], [6]. The relationship between the pathogen's available carbon and energy sources, or host nutritional niche [7], and pathways required for replication in vivo has been demonstrated for a variety of pathogenic microbes. Extraintestinal pathogenic E. coli require peptide import systems, the TCA cycle, and gluconeogenesis to consume amino acids and peptides in the urinary tract [8], while intestinal pathogenic E. coli require pathways to catabolize multiple sugars available in the intestine [9]. Salmonella enterica serovar Typhimurium exploits the inflammatory response of the host that creates an alternative electron acceptor to allow the pathogen to respire and compete with anaerobic gut residents [10]. The energetic consequences of modulating respiratory chain components and proton motive force can also promote pathogen survival in the face of bactericidal activities of the host [11].
The contribution of central carbon pathways to pathogenesis has been shown for numerous intracellular and extracellular nutritional niches occupied by pathogens. Expression-based and genetic approaches using the model cytosolic pathogen, Listeria monocytogenes, indicate that gluconeogenesis and uptake and catabolism of glycerol and dihydoxyacetone are required for bacterial replication in vivo [12], [13], [14], [15]. These findings are supported by the observation that disruption of glucose uptake has no effect on L. monocytogenes intracellular replication [16], suggesting that glycerol is the preferred carbon source for Listeria in vivo. Shigella flexneri, which also replicates in the host cell cytosol, requires glycerol-3-phosphate as a carbon source [17]. Interestingly, enteroinvasive E. coli (EIEC), which are genetically-related to S. flexneri, also use 3-carbon substrates as a carbon source in vivo. Because chorismate, GMP, and thymidylate synthesis have been found essential for S. flexneri replication in vivo [18], [19] and de novo synthesis of amino acids is extensive, it is believed that glycerol metabolism and anabolic pathways in this bacterium and EIEC may be important for bacteria that replicate within epithelial cells during intestinal infection.
Mycobacterium tuberculosis that replicates intracellularly within phagocytes, utilizes fatty acids in addition to glycerol or glycerol-3-phosphate as a carbon source in macrophages [20]. Seminal studies show that in vivo carbon metabolism in M. tuberculosis is dependent on fatty acid catabolism and the glyoxylate shunt through the TCA cycle [21], [22], [23]. The collective requirement for glycerol catabolism for L. monocytogenes, S. flexneri, EIEC, and M. tuberculosis growth in vivo likely indicates that glycerol is a readily available carbon source inside host cells and extends Rolf Freter's nutrient-niche hypothesis [7] to include available nutrients within a eukaryotic cell. However, despite occupying a similar host microenvironment, intracellular glycerol is not the preferred carbon source for S. enterica serovar Typhimurium because glucose import, glycolysis, and the oxidative TCA cycle are required for Salmonella to colonize the intestine and replicate within host phagocytes [24], [25], [26], [27]. This key difference in preferred carbon source in vivo could reflect a Salmonella fitness adaptation for facing increased competition with a diversity of luminal gut anaerobes.
Extracellular or luminal colonizers, including both commensal and pathogenic E. coli, are able to occupy the host gastrointestinal tract, yet their nutritional requirements for carbon metabolism in vivo have key differences. Colonization studies using the streptomycin-treated mouse model in combination with transcriptional profiling during culture in mucus demonstrated that the ED pathway, and gluconate or other sugar acids, are required for intestinal growth of commensal E. coli [28]. EHEC requires similar central metabolic pathways as commensal strains, however, EHEC colonization in vivo requires the catabolism of up to six additional sugars [9]. EHEC also utilizes glycolytic substrates and switches to gluconeogenic substrates when present in the intestine with commensal E. coli, which solely utilizes glycolytic pathways for in vivo growth [29]. This finding, that competition in vivo can alter preferred routes of carbon flux through the central pathways, introduces the notion that studying polymicrobial interactions during host colonization is essential to understand the relationship between bacterial metabolism and pathogenesis.
For extraintestinal pathogenic E. coli (ExPEC), it has been shown that D-serine metabolism and acetogenic growth are important during colonization of the urinary tract [30], [31]. In previous work, we have demonstrated that the import of peptides, gluconeogenesis, and the TCA cycle are required for E. coli during extraintestinal infection, while glycolysis and the pentose phosphate pathway are dispensable [8]. This indicates E. coli has to synthesize sugars from amino acids (gluconeogenesis) while enzymes for sugar catabolism have no affect on fitness. Although less is known about the in vivo metabolism of Proteus mirabilis, another important urinary tract pathogen, it would be expected to have the same enzymatic requirements during infection. Attenuated strains of P. mirabilis have been identified with mutations in genes that encode proteins involved in gluconate and pyruvate metabolism, and in enzymes of the TCA cycle, using signature-tagged mutagenesis [32], [33]. These earlier studies are supported by a recent comparison of global gene expression studies from E. coli [34], [35] and P. mirabilis [36] that indicated many similarities and some subtle differences may exist in vivo between these uropathogens during experimental infection of the urinary tract. To better understand the relationship between the host nutritional niche and pathogen growth, we used defined mutants, each defective in specific metabolic pathways, to directly examine the in vivo metabolism for two bacterial pathogens that occupy the same host niche and likely have access to the same nutrients during infection.
Unexpectedly, we found remarkably divergent in vivo requirements for central pathways between these two pathogens during UTI by assessing the in vivo fitness of strains containing mutations in pgi, pfkA, tpiA, pykA, gnd, talB, edd, sdhB, fumC, frdA, and pckA, in both uropathogenic E. coli CFT073 and P. mirabilis HI4320 (Fig. 1). Because the urinary tract is a normally sterile environment, in this current study, we not only further characterized the role of central metabolism during host colonization for both pathogenic E. coli and P. mirabilis in mono-species infection, but it was also possible to develop a polymicrobial infection model using the host urinary tract as an unoccupied vessel. Using this new model and, as their complementary utilization of central pathways suggested, we found that co-infection with E. coli and P. mirabilis wild-type strains enhanced bacterial colonization and persistence of both pathogens during UTI. These findings help explain the molecular and biochemical basis of polymicrobial infection in the urinary tract.

Results
Glycolysis is required for P. mirabilis and dispensable for E. coli during urinary tract infection
Despite extensive biochemical and in vitro studies of the model organism E. coli, characterization of central carbon pathways for extraintestinal pathogenic E. coli during infection is considerably less well understood than well-considered virulence factors [6]. Recently, a uropathogenic isolate was used to investigate pathogenic E. coli central metabolism in an infection model and found that in contrast to commensal E. coli, glycolysis is dispensable for extraintestinal pathogenic E. coli during colonization, while gluconeogenesis is required during infection [8]. One limitation from that study is the glycolytic enzymes investigated in that work could also play a role in gluconeogenesis. To address this, additional glycolytic mutants were constructed in E. coli CFT073, a prototype strain isolated from the blood and urine of a patient with acute pyelonephritis and urosepsis [37], [38]. In addition to strains lacking tpiA (triose phosphate isomerase) and pgi (phosphoglucose isomerase), mutants in irreversible glycolytic steps involving both 6-carbon (pfkA; 6-phosphofructokinase transferase) and 3-carbon (pykA; pyruvate kinase) substrates were constructed and tested in competitive infections with the parental E. coli CFT073 strain. Using the well-established murine model of ascending infection [39], we found that disruption of either the preparative or substrate level phosphorylation stages of glycolysis had no effect on the ability of E. coli to compete with wild-type CFT073 during experimental infection (Fig. 2A).

Because the growth medium within the urinary tract, urine, is a dilute mixture of amino acids and peptides [40], it is not unexpected that glycolysis would be dispensible during UTI. The composition of urine and its relative lack of available carbohydrates (except under diabetic condition), along with the lack of a significant contribution of glycolysis for E. coli during experimental UTI, predicts that glycolysis mutants in P. mirabilis, another common urinary tract pathogen, would have no apparent fitness defect in vivo. To test this, mutations were constructed in P. mirabilis HI4320, in the same glycolysis genes tested for E. coli: pgi, pfkA, tpiA, and pykA. Unexpectedly, any mutation that disrupted glycolysis in P. mirabilis resulted in a significant fitness defect using the same model of ascending infection (Fig. 2B). With the exception of pykA that demonstrated a fitness defect in the bladder (P = 0.031), all of the remaining P. mirabilis glycolysis mutants tested were out-competed by wild-type parental HI4320>1,000-fold (P<0.050) in both bladders and kidneys (Fig. 2B).
The dramatic differential requirement for glycolysis during infection between E. coli and P. mirabilis was not due to differences during in vitro growth. In both E. coli and P. mirabilis, only mutants in pfkA, pgi, and tpiA demonstrated the expected growth defect in defined medium containing glucose as the sole carbon source; growth rates of pykA mutants in defined medium containing glucose was similar to wild-type for both strains (Fig. 2C and D). Both sets of glycolysis mutants also demonstrated growth rates similar to the parental strains in LB medium (Fig. 2E and F). It was possible to complement the in vitro growth defect for the P. mirabilis tpiA mutant by introducing the tpiA gene from wild-type HI4320 into the mutant strain on a low copy plasmid (Fig. 2H). The re-introduction of the wild-type tpiA allele also restored the ability of the mutant strain to colonize the urinary tract; in vivo complementation resulted in a complete reversal of the tpiA fitness defect during competitive infection with the parental wild-type HI4320 harboring an empty vector control plasmid (Fig. 2I). It was also possible to heterologously complement the growth defect for the E. coli tpiA mutant in defined medium containing glucose by introduction of the wild-type tpiA allele cloned from P. mirabilis HI4320 (Fig. 2G).
E. coli and P. mirabilis transaldolase mutants have impaired fitness during UTI
Previously, it has been shown that mutation of E. coli transaldolase A gene (talA) does not negatively affect fitness during extraintestinal infection despite TalA being induced by CFT073 when cultured in human urine [8]. This suggests that the non-oxidative pentose phosphate pathway does not significantly contribute to pathogen fitness during urinary tract infection. To better characterize the contribution of the isomerizations of the non-oxidative pentose phosphate pathway in vivo, an additional transaldolase mutant, transaldolase B (talB), was constructed in E. coli CFT073. TalB is the major transaldolase in E. coli that transfers a three-carbon moiety from a C7 molecule to glyceraldehyde-3-P (C3) to form erythrose-4-P (C4) and fructose-P (C6). This stage of the pentose phosphate pathway is reversible, and thus, can be uncoupled from the oxidative decarboxylation reactions that produce NADPH. While loss of the major transaldolase, TalB, did not affect E. coli fitness during UTI (Fig. 3A); P. mirabilis talB mutant bacteria were out-competed>100-fold by wild-type in both the bladders and kidneys (P<0.003) (Fig. 3B).

One possibility for a difference between E. coli and P. mirabilis requirement in the non-oxidative pentose phosphate pathway is the redundancy of transaldolase in E. coli [41]. P. mirabilis strains encode a single transaldolase enzyme (TalB), while E. coli strains have both TalA and TalB, which catalyze identical reactions for the cell. To determine if the lack of a fitness defect for the E. coli talB mutant (Fig. 3A), is due to functional redundancy, we tested the talA single mutant and constructed and tested a talA talB double mutant strain in competitive infections with the parental CFT073 wild-type strain. In these studies, we found that lack of talA resulted in the wild-type being outcompeted by the single mutant in the bladder (P = 0.043) (Fig. 3C). Although not as striking as the talB mutant in P. mirabilis, loss of both talA and talB in E. coli resulted in the transaldolase double mutant being out-competed by wild-type CFT073>5.0-fold in bladders and kidneys (P<0.050) (Fig. 3D).
Disruption of the Entner-Doudoroff pathway or NADPH production in the pentose phosphate pathway creates a fitness defect in P. mirabilis but does not affect E. coli fitness during UTI
To distinguish the relative importance of the oxidative branch of the pentose phosphate pathway from the non-oxidative transaldolase-containing branch, a mutant in phophogluconolactonate (gnd) and a mutant defective in gluconate catabolism, 6-phosphoglyconate dehydrase (edd), were tested in competitive infections with the parental CFT073 wild-type strain. E. coli lacking either the oxidative pentose phosphate pathway (gnd), or the ED pathway (edd) were recovered from bladders and kidneys in numbers not significantly different from the wild-type strain (median CI = 1) (Fig. 3A). Surprisingly, unlike the lack of contribution for oxidative production of NADPH in the pentose phosphate pathway for E. coli, P. mirabilis mutants in gnd were out-competed>100-fold by wild-type in both the bladders and kidneys (P<0.003) (Fig. 3B).
Previously, signature-tagged mutagenesis identified a P. mirabilis edd transposon insertion as attenuated during experimental UTI [33], however, the attenuation caused by the disruption of edd was not confirmed by testing a ‘clean’ isogenic mutant strain in vivo. Despite this, it was reasonable to expect that, similar to the different requirement for glycolysis between E. coli and P. mirabilis during infection, P. mirabilis may require the capacity to metabolize gluconate via the Entner-Duodoroff pathway in vivo. Consistent with this, we found that in contrast to the findings with E. coli, the P. mirabilis edd mutant was significantly out-competed in both the bladders and kidneys by the parental HI4320 strain (P<0.020) during co-challenge infections when co-inoculated 1∶1 with wild-type (Fig. 3B). With the exception of talA, both E. coli and P. mirabilis share the same complement of the Entner-Duodoroff and pentose phosphate genes; therefore it is unlikely that redundancy of transaldolase in E. coli can account for the disparate requirements for these pathways between the two pathogens. In support of this, the edd, talA, talB, talAtalB, and gnd strains in E. coli and the edd, talB, and gnd strains in P. mirabilis all demonstrate similar growth in vitro and also to both wild-type parental strains during culture in LB medium and defined medium containing glucose as the sole carbon source (S1 Fig.).
In vivo contribution of the TCA cycle and gluconeogenesis during urinary tract infection
The aerobic tricarboxylic acid (TCA) cycle has been proposed to be required for E. coli fitness during growth on gluconeogenic substrates present in the urinary tract [8]. Specifically, E. coli sdhB mutant bacteria have been shown to have fitness defects during UTI [8], [42], suggesting that the reductive TCA cycle may not be operating during host colonization. To better define the role for the TCA cycle during extraintestinal infection, mutants of E. coli and P. mirabilis lacking succinate dehydrogenase; sdhB, fumarate dehydratase (fumarase); fumC, and fumarate reductase; frdA were constructed and tested in competitive infections with wild-type E. coli CFT073 or P. mirabilis HI4320, respectively. While both E. coli and P. mirabilis required TCA cycle reactions for fitness in vivo, sdhB was required for fitness only during cystitis (bladder CFU) in E. coli (Fig. 4A) and only during pyelonephritis (kidney CFU) in P. mirabilis (Fig. 4B) (P>0.050). It is generally believed that the production of reduced FADH2 during the conversion of succinate to fumarate by succinate dehydrogenase is avoided during fermentation by modification of the TCA cycle to an incomplete reductive pathway where fumarate conversion to succinate by fumarate reductase replaces succinate dehydrogenase activity. The loss of FrdA resulted in a fitness defect for P. mirabilis during infection of both the bladder and kidneys (P>0.005) (Fig. 4B). In contrast, E. coli frdA mutant colonization levels were indistinguishable from wild-type (median CI = 1.0) in the kidneys and significantly outcompeted the parental CFT073 strain>50-fold during acute cystitis (P = 0.024) (Fig. 4A). Both E. coli and P. mirabilis required fumC, which functions to convert fumarate to malate; loss of FumC, however, resulted in a severe fitness defect for P. mirabilis during bladder and kidney infection (CI<10−3, P<0.005), while the E. coli fumC mutant colonized the kidneys to similar levels as the parental CFT073 strain (CI = 0.94) and had a minor fitness defect in the bladder (CI = 0.1, P = 0.031) (Fig. 4A).

During bacterial growth on gluconeogenic substrates, peptides and certain amino acids that are present in the urinary tract are broken-down into pyruvate, which can be oxidized in the TCA cycle or reduced to fermentative end-products. The resulting oxaloacetate can fuel gluconeogenesis as the substrate for pyruvate carboxykinase (pckA) that generates phophoenolpyruvate and bypasses the irreversible glycolytic reaction catalyzed by pyruvate kinase (pykA). Mutation of pckA, which disrupts gluconeogenesis, resulted in a significant fitness defect for E. coli in both bladder and kidneys (P<0.005) (Fig. 4A). Loss of pckA in P. mirabilis did not significantly affect colonization during urinary tract infection (Fig.4B). In support of differential utilization of amino acids present in the urinary tract, arginine and serine auxotrophs of E. coli demonstrate no fitness defect during UTI [8], while in P. mirabilis, serine auxotrophy created a 100-fold decrease in bladder colonization (CI = 10−2, P<0.005) (S2 Fig.).
The variable requirement for gluconeogenesis between both pathogens was not due to differences during in vitro culture; pckA mutant strains in both CFT073 and HI4320 backgrounds were indistinguishable from parental strains in LB medium and defined medium containing glucose (Fig. 4C and D). In E. coli, the sdhB mutant demonstrated a growth defect in LB medium (Fig. 4C) but not when cultured in defined glucose medium (Fig. 4E), while the P. mirabilis frdA mutant demonstrated a growth defect in defined glucose medium (Fig. 4F) but not when cultured in LB medium (Fig. 4D). Both E. coli and P. mirabilis with mutations in fumC demonstrated an in vitro growth defect in LB medium, but only the P. mirabilis fumC mutant was unable to replicate in defined glucose medium (Fig. 4F). Although both of these pathogenic isolates require components of the TCA cycle for fitness during infection, these in vivo and in vitro data suggest a key difference in respiration during growth on glycolytic substrates exists between E. coli and P. mirabilis despite both being enteric bacteria.
Polymicrobial UTI shifts the fitness requirement for oxidative pentose phosphate from P. mirabilis to E. coli
The striking difference in the central pathway requirements during UTI between E. coli and P. mirabilis are puzzling because the central pathways are conserved and both pathogens are being assessed for fitness in the identical ascending UTI model. This suggests that an activity associated with the growth of the bacteria may cause alterations in the nutrient availability within the urinary tract. To test this, we performed mixed infections where the same mutation was tested against the opposite wild-type isolate. It was possible to distinguish E. coli from P. mirabilis by performing viable counts on agar with and without tetracycline due to P. mirabilis innate TetR phenotype. We chose to test the gnd mutants in both E. coli and P. mirabilis because that mutation created a severe fitness defect in P. mirabilis at 7 days in the bladder and kidneys, while no effect on fitness was observed for E. coli at 48 h in either tissue (Fig. 2A and B). In addition, the P. mirabilis gnd mutant demonstrated a fitness defect at 48 h in both the bladder and kidneys (Fig. 5A). Surprisingly, we found that when mixed 1∶1 with wild-type E. coli CFT073, the P. mirabilis gnd mutant was not out-competed at 48 h in either bladder or kidneys (Fig. 5B). Further, the E. coli gnd mutant was now significantly out-competed by the wild-type P. mirabilis HI4320 by>100-fold in both the bladder and kidneys (Fig. 5C). When the gnd mutants of each strain were mixed 1∶1, there was no observable difference in competitive indices (Fig. 5D). The same apparent reversal of in vivo fitness was also observed at 7 days post-inoculation (Fig. 5E), while no in vitro growth advantage was observed in any combination of gnd mutant bacteria and wild-type E. coli or P. mirabilis (Fig. 5F). Similarly, E. coli and P. mirabilis wild-type demonstrate equivalent growth during co-culture in LB medium and in defined medium with glucose as the sole carbon source (S3 Fig.).

Polymicrobial infection with E. coli and P. mirabilis enhances bacterial colonization of the urinary tract
The observed differences in central pathways requirements between E. coli and P. mirabilis during colonization of the urinary tract led us to speculate that both species could co-exist within the same urinary tract without directly competing for nutrients. By performing mixed inoculations of E. coli and P. mirabilis, it was also possible to test whether the observed fitness reversal of the gnd mutant bacteria could be explained by intrinsic differences in the level of colonization during UTI. Indeed, as expected, the CFU/g of bladder and kidneys at 7 d are 2–3 logs higher for P. mirabilis than E. coli during single strain infection (Fig. 6A and B). When the wild-type E. coli CFT073 and P. mirabilis HI4320 were co-inoculated, however, the level of E. coli colonization in the bladder and kidneys increased by over 3 logs at both 48 h and 7 days post-inoculation (Fig. 6C and D) with a concomitant 10-fold increase in P. mirabilis colonization in both the bladder and kidneys (Fig. 6D). To further test the compatibility of these uropathogens we also performed sequential infections. Mice were inoculated with a single strain and colonization was established for 48 h prior to infecting with the second strain. In these sequential infections we observed that pre-colonization of the urinary tract did not exclude colonization by the second strain (S4 Fig.). Further, when P. mirabilis is used to infect and colonize mice, followed by infection with E. coli we observed enhanced colonization as seen when both bacteria are simultaneously co-inoculated into mice (S4 Fig.). These data demonstrate that mixed infection provides an obvious benefit for E. coli during UTI, but also that the presence of E. coli provides a mutual benefit by allowing P. mirabilis to colonize to a greater density than it would by itself.

Discussion
The primary objective for microorganisms is to grow or replicate. For pathogenic microorganisms this need to replicate is paramount for their ability to successfully colonize, establish infection and cause disease. Many bacteria have evolved specific pathways that provide a growth advantage in a specific nutritional niche. These specific pathways often involve transport systems that aid the bacterium to acquire a certain nutrient, such as the ability of uropathogenic E. coli to import and utilize D-serine [31], [43] or the ability to sense α-ketoglutarate levels [44]. In contrast to these specific types of adaptations, most bacteria share highly conserved central pathways colloquially referred to as central metabolism. Our previous work began the comprehensive characterization of central pathways required by E. coli during UTI and found that gluconeogenesis and the TCA cycle were required to catabolize the dilute mixture of amino acids and peptides found in the urinary tract, while glycolysis, the pentose phosphate pathway, and the Entner Duodoroff pathway were dispensible [8]. In the present study, we extended this comprehensive study of E. coli central pathway requirements during UTI and in addition, performed parallel experiments using P. mirabilis, another well-studied uropathogen. We reasoned that both species of bacteria would have similar central metabolism requirements because they both occupy the same host niche, the urinary tract. Unexpectedly, we found that E. coli and P. mirabilis have strikingly divergent central pathway requirements despite infecting and growing in an identical host environment with presumably access to the same nutrients.
Our finding that E. coli and P. mirabilis have different central pathway requirements suggests that either a specific activity of the bacteria alters the host niche or that there is an intrinsic difference in the metabolic capabilities between the bacteria. Since we are studying highly conserved central pathways that are present in both species it is reasonable to conclude that a specific activity is present in one that is lacking in the other species. When considering the host urinary tract, the available nutrients are amino acids, peptides, and urea [40]. One obvious difference between Proteus and E. coli is that the former produces urease enzyme [45], [46], which hydrolyzes urea into ammonia and carbon dioxide (Fig. 7). The presence of urease activity would create a nitrogen rich environment by vastly increasing nitrogen availability via the concomitant increased ammonia concentration. In turn, the C/N ratio would be dramatically reduced for a urease producing bacteria like P. mirabilis relative to E. coli that does not have genes to encode urease. The altered C/N ratio would have profound effects on central pathway utilization because carbon metabolism and nitrogen assimilation is highly integrated [47], [48], [49]. Indeed, the apparent divergence in the ability to sense available nitrogen in urea results in E. coli activation of the glutamine synthetase and glutamate oxo-glutarate aminotransferase system (GS/GOGAT) to assimilate nitrogen [34], [35], while P. mirabilis assimilates nitrogen, via glutamate dehydrogenase (Gdh) [36] due to the apparent excess nitrogen available from ammonia produced by urea hydrolysis. We reason that this key difference is partly responsible for P. mirabilis requiring glycolysis, pentose phosphate pathway, and the ED pathway; while, on the other hand, the exact same mutations in E. coli have no affect on fitness during UTI. Alternatively, our findings raise the possibility that E. coli and P. mirabilis could reside in different cellular compartments. For example, it has been shown that E. coli can reside intracellularly during acute infection [50], [51], and while P. mirabilis can invade cultured cells [52], [53]; it is unclear if P. mirabilis spends a significant portion of the acute infection within host cells.
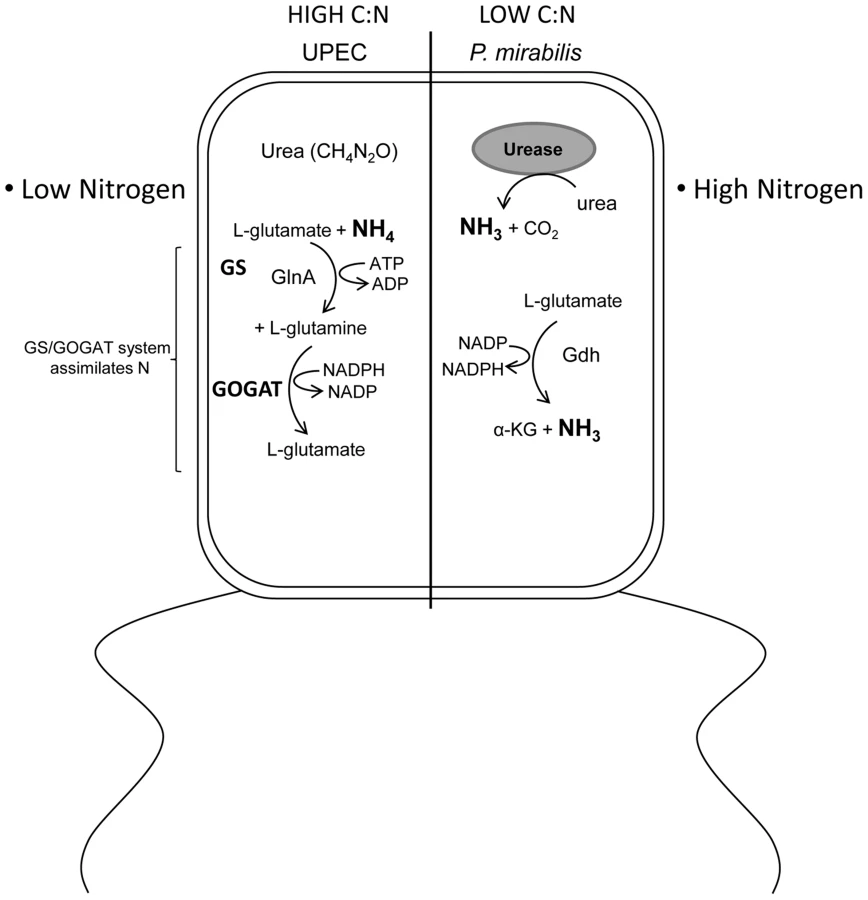
In addition to finding a remarkable difference in central metabolism requirements between two pathogens that infect the urinary tract, these findings have validated that glycolysis is dispensable for E. coli during UTI. In our previous work we assessed glycolysis by studying a mutation in the gene that encodes triose phosphate isomerase [8]; however, that enzyme is reversible. In the present study we created and tested phophofructokinase - and pyruvate kinase-deficient mutants in E. coli and P. mirabilis. These enzymes perform irreversible steps in glycolysis, thus by testing these mutations during UTI it is now clear that glycolysis is dispensible for E. coli and is required for P. mirabilis fitness in vivo. That glycolysis is required for P. mirabilis but not E. coli raises the possibility that sugars become available within the urinary tract during Proteus infection. That sugars become available when P. mirabilis colonizes the urinary tract would also allow for enhanced colonization of E. coli because sugars available during co-infection might increase growth of E. coli over numbers it would normally reach by solely consuming amino acids. This could also explain why gnd mutant E. coli displayed a fitness defect only when co-colonized with P. mirabilis.
It has also been shown that the oxidative TCA cycle is important for E. coli fitness during UTI [8], [42], but whether or not the reductive TCA cycle is important for fitness during UTI remains unanswered. We found that mutants lacking fumarate reductase, which is an enzyme that utilizes fumarate as an electron acceptor during anaerobic respiration, do not have a fitness defect in E. coli as predicted by our earlier study. In contrast, fumarate reductase mutants in P. mirabilis are out-competed by wild-type. These data show that the branched, reductive TCA pathway is not important for E. coli to grow within the urinary tract, supporting the notion that the urinary tract is moderately oxygenated [35]. However, the result that fumarate reductase is important for P. mirabilis fitness during UTI suggests an anaerobic environment does exist in the host urinary tract. In addition, because sensing oxygen depletion has been shown to be important for E. coli fitness during UTI [44], it remains possible that fumarate reductase is dispensible for E. coli due to the presence of alternative energy pathways like Ni-Fe hydrogenases and nitrate reductase, these and other redox enzymes are part of the large repertoire of electron transfer components available for the highly modular respiratory chains that can be assembled in E. coli [54]. Future studies will be useful to determine the exact energy pathways used by E. coli during UTI.
The urogenital track of humans is considered a site of polymicrobial colonization [55] and it has been shown that UTI can be polymicrobial [56], including co-infections with E. coli and P. mirabilis [57], [58]. Polymicrobial UTI has also been suggested to enhance E. coli virulence [58]. Our findings that two preeminent UTI pathogens have divergent central metabolism requirements have important implications for polymicrobial infections. Specifically, these data indicate that E. coli and P. mirabilis may not directly compete for nutrients during colonization of the urinary tract and led us to hypothesize that co-inoculation of E. coli and P. mirabilis may lead to enhanced colonization levels during experimental UTI. Indeed, we found that colonization was enhanced for both E. coli and P. mirabilis. The benefit of polymicrobial infection was more apparent for E. coli over time. In addition to supporting that E. coli and P. mirabilis may not directly compete for resources during UTI, these findings could alternatively suggest that some activity of one or both pathogenic bacteria may alter the host niche in such a way that facilitates bacterial replication. In support of this, we observed that P. mirabilis causes more tissue damage and larger induction of the pro-inflammatory cytokines IL-1α, IL-β, IL-6, and G-CSF than does E. coli in independent infection, while a mixed infection appears more similar to E. coli alone (S5 Fig.). With a systematic view of bacterial metabolism during UTI in hand, further studies of the host innate response to these infections will help form a comprehensive view of how the host response shapes bacterial carbon utilization during UTI.
To test whether or not the bacteria differences in the host urinary tract niche, we decided to co-inoculate E. coli and P. mirabilis mutants with the heterologous wild-type strain and assess fitness during UTI. Our findings showed that polymicrobial infection of the urinary tract changed the fitness results when compared to mono-species infection. The E. coli gnd mutant, which was not outcompeted by its parent strain, demonstrated a colonization disadvantage when co-inoculated with P. mirabilis wild-type. Conversely, the P. mirabilis gnd mutant, which was out-competed by its parent strain, did not demonstrate a disadvantage in colonization when co-inoculated with wild-type E. coli. Although future studies with additional mutants will be useful to further elucidate how co-inoculation might change central pathway requirements, these data do suggest that the host niche environment is altered by bacterial activity during UTI. Importantly, our study leads to a more complete picture of the metabolism of two key bacterial pathogens that cause UTI and shows that polymicrobial infection of the urinary tract may alter the metabolic pathways required for optimal growth within the host. These findings provide a better understanding of bacterial metabolism during clinically relevant infections and represent an important foundation to begin to dissect how metabolism and virulence intersect during UTI and how polymicrobial interactions may affect pathogenesis of extraintestinal E. coli infections.
Materials and Methods
Bacterial strains and culture conditions
P. mirabilis HI4320 was isolated from urine of a patient presenting with bacteriuria during long-term catheterization [56], [59]. E. coli CFT073 was isolated from the blood and urine of a patient with acute pyelonephritis [37]. E. coli and P. mirabilis were routinely cultured in lysogeny broth (LB) medium. For growth experiments, wild-type and mutant strains of E. coli and P. mirabilis were cultured in MOPS defined medium [60] and minimal salts medium [61], respectively, both containing either 0.2% (w/v) glucose or 0.2% (w/v) glycerol as the sole carbon source. Defined medium cultures were inoculated 1∶50 and LB cultures were inoculated 1∶100 from overnight LB bacterial cultures and incubated with aeration at 37°C. Growth curves were performed in triplicate; OD600 was recorded every hour.
Construction of metabolism mutants
P. mirabilis HI4320 mutants (S1 Table) were generated using the TargeTron Gene Knockout System (Sigma). Oligonucleotides for mutant construction were created using the TargeTron Design Site (Sigma). PCR confirmation of mutants was performed using oligonucleotides flanking the intron insertion site designed with the PrimerQuest program on the Integrated DNA Technologies website. E. coli CFT073 deletion mutants (S1 Table) were constructed using the lambda red recombinase system [62]. Primers homologous to sequences within the 5′ and 3′ ends of the target genes were designed and used to replace target genes with a nonpolar kanamycin - or chloramphenicol-resistance cassette derived from the template plasmid pKD4 or pKD3, respectively [62]. Confirmation of E. coli mutants was carried out by PCR using primers flanking the target gene sequence and comparing product size to wild-type PCR product size. When size differences were negligible PCR products were digested with a restriction enzyme (New England Biolabs).
Complementation of mutants
For in vitro complementation, the tpiA gene was amplified from P. mirabilis genomic DNA using Easy-A high fidelity polymerase (Stratagene) and independently cloned into pGEN-MCS [63], [64] using appropriate restriction enzymes. The sequences of pGEN-tpiA were verified by DNA sequence analysis prior to electroporation into the P. mirabilis tpiA mutant strain and E. coli tpiA mutant strain.
Experimental UTI
The CBA mouse model of ascending UTI [39], [65] was used to assess the fitness contribution of each metabolic mutant during co-challenge competition. To determine persistence of wild-type strains, independent infections of a single strain were performed. Female CBA/J mice (6–8 week old; 20 to 22 g; Jackson Laboratories) were anesthetized with ketamine/xylazine and transurethrally inoculated with a 50 µl bacterial suspension (total inoculum = 5×107 or 2×108 CFU) per mouse using a sterile polyethylene catheter (I.D. 0.28 mm × O.D. 0.61 mm) connected to an infusion pump (Harvard Apparatus). For in vivo co-challenges, a suspension containing 5×107 CFU of a 1∶1 ratio of P. mirabilis HI4320 and P. mirabilis kanamycin-resistant mutant in LB medium or a suspension containing 2×108 CFU of a 1∶1 ratio of E. coli CFT073 and E. coli antibiotic-resistant mutant in PBS. For independent infections, the respective P. mirabilis or E. coli suspensions contained only the wild-type strain. Input CFU/ml was determined by plating serial dilutions (Spiral Biotech) of each inoculum onto low salt (0.5 g NaCl/L) LB agar, to prevent P. mirabilis swarming, with and without antibiotic. For experiments with P. mirabilis, low salt LB agar (0.5 g NaCl/L) was used to prevent swarming. Infected mice were euthanized 48 h or 7 d post infection, bladder and kidneys were aseptically removed, weighed, and homogenized (OMNI International) in 3 ml PBS, and appropriate dilutions were spiral plated on LB agar with and without antibiotic to determine the output CFU/g of tissue. Viable counts were enumerated using QCount software (Spiral Biotech) and CFU from antibiotic-containing medium (mutant CFU) were subtracted from the total CFU from plates lacking antibiotic to determine the number of wild-type bacteria. For co-challenge experiments, competitive indices (CI) were calculated by dividing the ratio of the CFU of mutant to wild-type recovered from each mouse following infection by the ratio of the CFU of mutant to the CFU of wild-type present in the input. CI data were log-transformed and analyzed by the Wilcoxon signed-rank test to determine statistically significant differences in colonization (P-value <0.05). A CI>1 indicates that the mutant out-competes the wild-type strain and a CI<1 indicates that the mutant is out-competed by the wild-type strain. For independent infections the Mann-Whitney test was used to determine statistically significant differences in colonization (P-value <0.05).
Polymicrobial infections
The relative fitness in vivo for bacteria during polymicrobial infection was determined by co-inoculating UPEC CFT073 and P. mirabilis HI4320 strains and deletion mutants into the same female CBA mice as described previously with the following modification. For polymicrobial co-challenge infections, a 1∶1 (v/v) mixture was prepared containing 2.5×107 CFU of P. mirabilis HI4320 in LB medium and 108 CFU E. coli CFT073 in PBS. Competitive indices were calculated as described above. For polymicrobial infections containing only wild-type strains, the CFU/g tissue were determined following plating of serial dilutions on low salt (0.5 g NaCl/L) LB agar with and without tetracycline; P. mirabilis is intrinsically tetracycline-resistant. CFU from tetracycline agar plates, which represent P. mirabilis, were subtracted from total CFU recovered on LB agar without antibiotics to determine CFU/g for E. coli. For quantification of bacteria recovered from a polymicrobial infection with a wild-type strain and a heterologous kanamycin-resistant mutant strain, CFU on LB agar containing kanamycin (mutant) were subtracted from total CFU recovered on LB without antibiotics, to determine wild-type CFU/g tissue. Tetracycline was used to enumerate bacterial colonization levels following polymicrobial infection with heterologous kanamycin-resistant strains.
Ethics statement
All animal experiments were performed in accordance to the protocol (08999-3) approved by the University Committee on Use and Care of Animals at the University of Michigan. This protocol is in complete compliance with the guidelines for humane use and care of laboratory animals mandated by the National Institutes of Health.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. EisenreichW, DandekarT, HeesemannJ, GoebelW (2010) Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 8 : 401–412.
2. SomervilleGA, ProctorRA (2009) At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol Mol Biol Rev 73 : 233–248.
3. PoncetS, MilohanicE, MazeA, Nait AbdallahJ, AkeF, et al. (2009) Correlations between carbon metabolism and virulence in bacteria. Contrib Microbiol 16 : 88–102.
4. RohmerL, HocquetD, MillerSI (2011) Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19 : 341–348.
5. SmithH (2000) Questions about the behaviour of bacterial pathogens in vivo. Philos Trans R Soc Lond B Biol Sci 355 : 551–564.
6. AlteriCJ, MobleyHL (2012) Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr Opin Microbiol 15 : 3–9.
7. FreterR, BricknerH, BotneyM, ClevenD, ArankiA (1983) Mechanisms that control bacterial populations in continuous-flow culture models of mouse large intestinal flora. Infect Immun 39 : 676–685.
8. AlteriCJ, SmithSN, MobleyHL (2009) Fitness of Escherichia coli during urinary tract infection requires gluconeogenesis and the TCA cycle. PLoS Pathog 5: e1000448.
9. FabichAJ, JonesSA, ChowdhuryFZ, CernosekA, AndersonA, et al. (2008) Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun 76 : 1143–1152.
10. WinterSE, ThiennimitrP, WinterMG, ButlerBP, HusebyDL, et al. (2010) Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467 : 426–429.
11. AlteriCJ, LindnerJR, ReissDJ, SmithSN, MobleyHL (2011) The broadly conserved regulator PhoP links pathogen virulence and membrane potential in Escherichia coli. Mol Microbiol 82 : 145–163.
12. EylertE, ScharJ, MertinsS, StollR, BacherA, et al. (2008) Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol Microbiol 69 : 1008–1017.
13. JosephB, MertinsS, StollR, ScharJ, UmeshaKR, et al. (2008) Glycerol metabolism and PrfA activity in Listeria monocytogenes. J Bacteriol 190 : 5412–5430.
14. JosephB, PrzybillaK, StuhlerC, SchauerK, SlaghuisJ, et al. (2006) Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol 188 : 556–568.
15. ChatterjeeSS, HossainH, OttenS, KuenneC, KuchminaK, et al. (2006) Intracellular gene expression profile of Listeria monocytogenes. Infect Immun 74 : 1323–1338.
16. StollR, GoebelW (2010) The major PEP-phosphotransferase systems (PTSs) for glucose, mannose and cellobiose of Listeria monocytogenes, and their significance for extra - and intracellular growth. Microbiology 156 : 1069–1083.
17. LucchiniS, LiuH, JinQ, HintonJC, YuJ (2005) Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect Immun 73 : 88–102.
18. NoriegaFR, LosonskyG, LauderbaughC, LiaoFM, WangJY, et al. (1996) Engineered deltaguaB-A deltavirG Shigella flexneri 2a strain CVD 1205: construction, safety, immunogenicity, and potential efficacy as a mucosal vaccine. Infect Immun 64 : 3055–3061.
19. CersiniA, MartinoMC, MartiniI, RossiG, BernardiniML (2003) Analysis of virulence and inflammatory potential of Shigella flexneri purine biosynthesis mutants. Infect Immun 71 : 7002–7013.
20. SchnappingerD, EhrtS, VoskuilMI, LiuY, ManganJA, et al. (2003) Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med 198 : 693–704.
21. McKinneyJD, Honer zu BentrupK, Munoz-EliasEJ, MiczakA, ChenB, et al. (2000) Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406 : 735–738.
22. Munoz-EliasEJ, UptonAM, CherianJ, McKinneyJD (2006) Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol 60 : 1109–1122.
23. Munoz-EliasEJ, McKinneyJD (2005) Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11 : 638–644.
24. Tchawa YimgaM, LeathamMP, AllenJH, LauxDC, ConwayT, et al. (2006) Role of gluconeogenesis and the tricarboxylic acid cycle in the virulence of Salmonella enterica serovar Typhimurium in BALB/c mice. Infect Immun 74 : 1130–1140.
25. Mercado-LuboR, GaugerEJ, LeathamMP, ConwayT, CohenPS (2008) A Salmonella enterica serovar typhimurium succinate dehydrogenase/fumarate reductase double mutant is avirulent and immunogenic in BALB/c mice. Infect Immun 76 : 1128–1134.
26. Mercado-LuboR, LeathamMP, ConwayT, CohenPS (2009) Salmonella enterica serovar Typhimurium mutants unable to convert malate to pyruvate and oxaloacetate are avirulent and immunogenic in BALB/c mice. Infect Immun 77 : 1397–1405.
27. BowdenSD, RowleyG, HintonJC, ThompsonA (2009) Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar typhimurium. Infect Immun 77 : 3117–3126.
28. ChangDE, SmalleyDJ, TuckerDL, LeathamMP, NorrisWE, et al. (2004) Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A 101 : 7427–7432.
29. MirandaRL, ConwayT, LeathamMP, ChangDE, NorrisWE, et al. (2004) Glycolytic and gluconeogenic growth of Escherichia coli O157: H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect Immun 72 : 1666–1676.
30. AnforaAT, HalladinDK, HaugenBJ, WelchRA (2008) Uropathogenic Escherichia coli CFT073 is adapted to acetatogenic growth but does not require acetate during murine urinary tract infection. Infect Immun 76 : 5760–5767.
31. AnforaAT, HaugenBJ, RoeschP, RedfordP, WelchRA (2007) Roles of serine accumulation and catabolism in the colonization of the murine urinary tract by Escherichia coli CFT073. Infect Immun 75 : 5298–5304.
32. BurallLS, HarroJM, LiX, LockatellCV, HimpslSD, et al. (2004) Proteus mirabilis genes that contribute to pathogenesis of urinary tract infection: identification of 25 signature-tagged mutants attenuated at least 100-fold. Infect Immun 72 : 2922–2938.
33. HimpslSD, LockatellCV, HebelJR, JohnsonDE, MobleyHL (2008) Identification of virulence determinants in uropathogenic Proteus mirabilis using signature-tagged mutagenesis. J Med Microbiol 57 : 1068–1078.
34. HaganEC, LloydAL, RaskoDA, FaerberGJ, MobleyHL (2010) Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog 6: e1001187.
35. SnyderJA, HaugenBJ, BucklesEL, LockatellCV, JohnsonDE, et al. (2004) Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect Immun 72 : 6373–6381.
36. PearsonMM, YepA, SmithSN, MobleyHL (2011) Transcriptome of Proteus mirabilis in the Murine Urinary Tract: Virulence and Nitrogen Assimilation Gene Expression. Infect Immun 79 : 2619–2631.
37. MobleyHL, GreenDM, TrifillisAL, JohnsonDE, ChippendaleGR, et al. (1990) Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 58 : 1281–1289.
38. WelchRA, BurlandV, PlunkettG3rd, RedfordP, RoeschP, et al. (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA 99 : 17020–17024.
39. HagbergL, EngbergI, FreterR, LamJ, OllingS, et al. (1983) Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infect Immun 40 : 273–283.
40. BrooksT, KeevilCW (1997) A simple artificial urine for the growth of urinary pathogens. Lett Appl Microbiol 24 : 203–206.
41. SprengerGA (1995) Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch Microbiol 164 : 324–330.
42. HadjifrangiskouM, KostakiotiM, ChenSL, HendersonJP, GreeneSE, et al. (2011) A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol Microbiol 80 : 1516–1529.
43. RoeschPL, RedfordP, BatcheletS, MoritzRL, PellettS, et al. (2003) Uropathogenic Escherichia coli use d-serine deaminase to modulate infection of the murine urinary tract. Mol Microbiol 49 : 55–67.
44. CaiW, WannemuehlerY, Dell'annaG, NicholsonB, BarbieriNL, et al. (2013) A novel two-component signaling system facilitates uropathogenic Escherichia coli's ability to exploit abundant host metabolites. PLoS Pathog 9: e1003428.
45. JonesBD, LockatellCV, JohnsonDE, WarrenJW, MobleyHL (1990) Construction of a urease-negative mutant of Proteus mirabilis: analysis of virulence in a mouse model of ascending urinary tract infection. Infect Immun 58 : 1120–1123.
46. JonesBD, MobleyHL (1988) Proteus mirabilis urease: genetic organization, regulation, and expression of structural genes. J Bacteriol 170 : 3342–3349.
47. MaoXJ, HuoYX, BuckM, KolbA, WangYP (2007) Interplay between CRP-cAMP and PII-Ntr systems forms novel regulatory network between carbon metabolism and nitrogen assimilation in Escherichia coli. Nucleic Acids Res 35 : 1432–1440.
48. SchumacherJ, BehrendsV, PanZ, BrownDR, HeydenreichF, et al. (2013) Nitrogen and carbon status are integrated at the transcriptional level by the nitrogen regulator NtrC in vivo. MBio 4: e00881–00813.
49. De LayN, GottesmanS (2009) The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 191 : 461–476.
50. MulveyMA, Lopez-BoadoYS, WilsonCL, RothR, ParksWC, et al. (1998) Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282 : 1494–1497.
51. MulveyMA, SchillingJD, MartinezJJ, HultgrenSJ (2000) Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A 97 : 8829–8835.
52. AllisonC, ColemanN, JonesPL, HughesC (1992) Ability of Proteus mirabilis to invade human urothelial cells is coupled to motility and swarming differentiation. Infect Immun 60 : 4740–4746.
53. MobleyHL, BelasR, LockatellV, ChippendaleG, TrifillisAL, et al. (1996) Construction of a flagellum-negative mutant of Proteus mirabilis: effect on internalization by human renal epithelial cells and virulence in a mouse model of ascending urinary tract infection. Infect Immun 64 : 5332–5340.
54. UndenG, BongaertsJ (1997) Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta 1320 : 217–234.
55. BrogdenKA, GuthmillerJM, TaylorCE (2005) Human polymicrobial infections. Lancet 365 : 253–255.
56. WarrenJW, TenneyJH, HoopesJM, MuncieHL, AnthonyWC (1982) A prospective microbiologic study of bacteriuria in patients with chronic indwelling urethral catheters. J Infect Dis 146 : 719–723.
57. SaverinoD, SchitoAM, ManniniA, PencoS, BassiAM, et al. (2011) Quinolone/fluoroquinolone susceptibility in Escherichia coli correlates with human polymicrobial bacteriuria and with in vitro interleukine-8 suppression. FEMS Immunol Med Microbiol 61 : 84–93.
58. CroxallG, WestonV, JosephS, ManningG, CheethamP, et al. (2011) Increased human pathogenic potential of Escherichia coli from polymicrobial urinary tract infections in comparison to isolates from monomicrobial culture samples. J Med Microbiol 60 : 102–109.
59. MobleyHL, WarrenJW (1987) Urease-positive bacteriuria and obstruction of long-term urinary catheters. J Clin Microbiol 25 : 2216–2217.
60. NeidhardtFC, BlochPL, SmithDF (1974) Culture medium for enterobacteria. J Bacteriol 119 : 736–747.
61. BelasR, ErskineD, FlahertyD (1991) Transposon mutagenesis in Proteus mirabilis. J Bacteriol 173 : 6289–6293.
62. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
63. GalenJE, NairJ, WangJY, WassermanSS, TannerMK, et al. (1999) Optimization of plasmid maintenance in the attenuated live vector vaccine strain Salmonella typhi CVD 908-htrA. Infect Immun 67 : 6424–6433.
64. LaneMC, AlteriCJ, SmithSN, MobleyHL (2007) Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc Natl Acad Sci U S A 104 : 16669–16674.
65. JohnsonDE, LockatellCV, Hall-CraigsM, MobleyHL, WarrenJW (1987) Uropathogenicity in rats and mice of Providencia stuartii from long-term catheterized patients. J Urol 138 : 632–635.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2015 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation