
Chronicky zvýšená teplota či horečka s nevelkou lymfadenopatií může být Castlemanova choroba, pokud se neprokáže maligní, autoimunitní anebo infekční příčina lymfadenopatie
Chronic low-grade fever or fever with mild lymphadenopathy may indicate Castleman disease if no malignant, autoimmune, or infectious cause of the lymphadenopathy is identified
Background: Diseases caused by immune system disorders fall under the care of every medical specialty. In hematology, these are diseases caused by autoantibodies, several autoinflammatory diseases (Schnitzler syndrome and VEXAS syndrome), as well as immune disorder-induced diseases presenting with systemic inflammatory reactions and lymphadenopathy. In the 5th edition of the WHO classification, these diseases are categorized under the chapter “Tumour-like lesions with B-cell predominance”, which includes Castleman disease (CD). Objective: This paper provides an overview of information on CD from the perspective of the years 2025 and 2026. In patients with unicentric CD (UCD), associations with paraneoplastic pemphigus or obliterative bronchiolitis have been described in rare cases. Prognostically unfavorable is the transformation of UCD into a follicular dendritic cell sarcoma. In cases of multicentric CD (MCD), the etiology is exceptionally identified as an infection with human herpesvirus 8. In rare cases of MCD combined with POEMS syndrome, the etiology is considered to be a monoclonal gammopathy. In most cases of MCD, none of the aforementioned etiologies are demonstrated, and these forms of MCD are referred to as idiopathic MCD (iMCD). The most aggressive form of iMCD is called iMCD-TAFRO, and its course resembles a cytokine storm. The form with the lowest aggressiveness was defined recently – idiopathic plasmacytic lymphadenopathy (iMCD-IPL) with a high concentration of polyclonal immunoglobulins and a higher concentration of the IgG4 subclass of immunoglobulins. Between these two extreme forms lies the majority of cases falling into the iMCD-NOS (not otherwise specified) category. Differences in the aggressiveness are caused by differences in etiopathogenesis. The text presents recent criteria for these diseases, information on symptoms, diagnosis, and prognosis. Conclusion: In MCD, suspicion must be raised in individuals with a systemic inflammatory response and lymphadenopathy after ruling out autoimmune, malignant, and infectious etiologies. Diagnosis of CD is clinico-pathological, meaning it arises based on the exchange of information between the clinician, who must suspect the disease, and the pathologist, who examines the histomorphological findings while taking into account the information provided by the clinicians.
Keywords:
Castleman disease
Autori:
prof. MUDr. Adam Zdeněk, CSc. 1; MUDr. Kamarádová Kateřina, Ph.D. 2; doc. MUDr. Doubková Martina, Ph.D. 3; doc. MUDr. Řehák Zdeněk, Ph.D. 4; MUDr. Koukalová Renata, Ph.D. 4; MUDr. Křivanová Andrea, Ph.D. 1; doc. MUDr. Horváth Teodor, Ph.D. 5; MUDr. Čermák Aleš, Ph.D. 6; MUDr. Boichuk Ivanna 1; MUDr. Štork Martin, Ph.D. 1; MUDr. Sandecká Viera, Ph.D. 1; MUDr. Adamová Zuzana, Ph.D. 7; prof. MUDr. Pour Luděk, Ph.D. 1
Pôsobisko autorov:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Fingerlandův ústav patologie, LF UK v Hradci Králové
2; Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
3; Oddělení nukleární medicíny, MOU Brno
4; Chirurgická klinika LF MU a FN Brno
5; Urologická klinika LF MU a FN Brno
6; Chirurgické oddělení, Moravskoslezská nemocnice Frýdek-Místek
7
Vyšlo v časopise:
Klin Onkol 2026; 39(3): 169-183
Kategória:
Přehled
doi:
https://doi.org/10.48095/ccko2026169
Súhrn
Východiska: Do péče každého medicínského oboru spadají choroby způsobené poruchou imunity. V hematologii jsou to autoimunitní nemoci způsobené autoprotilátkami, několik chorob autoinflamatorních (syndrom Schnitzlerové a VEXAS syndrom) a také choroby vyvolané poruchou imunity s projevy systémové zánětlivé reakce a s lymfadenopatií. V 5. verzi WHO klasifikaci jsou tyto nemoci řazeny do kapitoly Tumour-like lesions with B-cell predominance a patří mezi ně Castlemanova choroba (CD). Cíl: Práce přináší přehled informací o CD z pohledu let 2025–2026. U pacientů s unicentrickou CD (UCD) byla v ojedinělých případech popsána asociace s paraneoplastickým pemfigem anebo s obliterující bronchiolitidou. Prognosticky nepříznivá je transformace UCD do sarkomu z folikulárních dendritických buněk. V případech multicentrické CD (MCD) se výjimečně podaří prokázat etiologii v infekci humánním herpetickým virem 8. Ve vzácných případech kombinace MCD s POEMS syndromem je etiologie spatřována v monoklonální gamapatii. U většiny případů MCD se ani jedna z uvedených etiologií neprokáže a tyto formy MCD se nazývají idiopatická MCD (iMCD). Nejagresivnější forma iMCD se nazývá iMCD-TAFRO, svým průběhem připomíná cytokinovou bouři. Nejméně agresivní forma se nazývá iMCD-IPL (idiopatická plazmocytární lymfadenopatie). Vyznačuje se vysokou koncentrací polyklonálních imunoglobulinů a vyšší koncentrací podtřídy imunoglobulinu IgG4. A mezi těmito dvěma krajními formami se nalézá většina případů spadajících do kategorie blíže nespecifikovaná iMCD – iMCD-NOS (not otherwise specified.) Rozdíly v agresivitě jsou způsobeny rozdílnou etiopatogenezí. Text přináší recentní kritéria těchto chorob, informace o příznacích, diagnostice a o prognóze. Závěr: Na MCD je nutno pojmout podezření u osob se systémovou zánětlivou reakcí a lymfadenopatií po vyloučení autoimunitní, maligní a infekční etiologie. Diagnóza CD je klinicko-patologická neboli vzniká na základě výměny informací mezi klinikem, který na tuto chorobu musí pojmout podezření, a patologem, který se na histomorfologický nález dívá se zohledněním informací od kliniků.
Klíčová slova:
Castlemanova choroba
Úvod
Zařazení Castelmanovy choroby
Každý obor medicíny má na starosti některé pro daný obor specifické nemoci, jejichž etiopatogeneze spočívá v poruše určité složky imunity. Do spektra chorob, o něž pečují hematologové, spadá více chorob, jejichž příčinou je narušená imunita. Jsou to choroby způsobené autoprotilátkami (autoimunitní cytopenie aj.), patří sem i několik chorob autoinflamatorních (VEXAS syndrom, syndrom Schnitzlerové). Někdy dominují hematologické symptomy u dalších autoinflamatorních chorob, SAPHO syndromu a septické formy Stillovy choroby dospělých [1]. Do skupiny hematologických nemocí způsobených poruchou imunity patří také několik chorob s chronickou systémovou zánětlivou reakcí a lymfadenopatií.
Jednou z nich je Castlemanova choroba (Castleman disease – CD), která je již zařazena do 5. verze WHO klasifikace krevních chorob. Je uvedena v kapitole nazvané Tumour-like lesions with B-cell predominance [2]. Ta obsahuje následující jednotky, u nichž ponecháme anglické termíny této klasifikace:
- reactive B-cell-rich lymphoid proliferations that can mimic lymphoma;
- IgG4-related disease;
- unicentric Castleman disease;
- idiopathic multicentric Castleman disease;
- HHV-8 associated multicentric Castleman disease.
CD je také uvedena v 10. verzi mezinárodní klasifikace nemocí. Je zařazena do skupiny nazvané Other neoplasmas of uncertain or unknown behavior of lymphoid, hematopoietic and related tissue a byl jí přidělen kód D47.Z2.
Pro stanovení diagnózy iMCD je zapotřebí multioborové spolupráce
Pro stanovení diagnózy iMCD má zásadní úlohu patologie, bez odpovídajícího histomorfologického nálezu nelze diagnózu stanovit. Za nejpřínosnější odběr je považována biopsie uzliny nejvíce akumulující fluorodeoxyglukózu (FDG) a na to je zapotřebí lékařů operačních oborů, hrudního či břišního chirurga a případně urologa, pokud je infiltrát v oblasti retroperitonea a pánve. K diferenciální diagnostice chronického kašle pacientů s iMCD je třeba přizvat plicního specialistu, který může stanovit diagnózu lymfocytární pneumonie anebo obliterující bronchiolitidy, dvou neobvyklých plicních chorob, které mohou iMCD provázet. Možné poškození ledviny lze verifikovat pouze biopsií a případné puchýřnaté projevy nutno zkonzultovat s dermatologem, který může potvrdit paraneoplastický pemfigus.
Velmi často jsou tito nemocní směrováni do revmatologických ambulancí s podezřením na autoimunitní chorobu, takže i revmatologové ji mohou diagnostikovat [3]. Klasifikace sice řadí nemoc mezi krevní choroby, ale bez multioborové spolupráce není přesné stanovení její diagnózy možné.
Stanovení diagnózy MCD
MCD na sebe upozorní dlouhodobými zánětlivými projevy se zvýšenou teplotou do 38 °C nebo horečkami > 38 °C. Lymfadenopatie je u MCD nevelká, průměr lymfatických uzlin je zvětšen jen na dvojnásobek či trojnásobek, takže obvykle mají průměr 2–4 cm. Lymfadenopatie může při prvním vyšetření uniknout a upozorní na ni až zobrazovací vyšetření.
Lékař prvního kontaktu konstatuje u osob MCD zánětlivou reakci se zvýšenou hodnotou C-reaktivního proteinu (CRP), případně s dalšími zvýšenými markery zánětu (feritinu, fibrinogenu) a také s anemií a trombocytózou. Tyto projevy mají mnoho možných vysvětlení. Nejdříve se obvykle pomýšlí na infekční příčinu a pacient dostane antibiotika. Když ta nepomohou, začne vyšetřující lékař přemýšlet o autoimunitním či autoinflamatorním onemocnění. Když základní vyšetření neobjasní diagnózu, tak jsou potíže u pacienta klasifikovány jako horečka nejasné etiologie (fever of unknown origin – FUO).
Klinickou jednotku FUO považujeme stále za velmi užitečnou, protože definuje skupinu nemocných, u nichž není vhodné dělat pokus s antibiotiky a odkládat hledání příčiny. Pokud je zřejmé, že pacient splnil kritéria FUO, musí se ihned začít s vyšetřováním etiologie.
FUO má časové kritérium – nejméně 3týdenní trvání horeček nebo subfebrilií. Za 3 týdny totiž většina samoodeznívajících chorob (self limiting disease), jako jsou virózy, angíny, vymizí, zatímco při závažném onemocnění (maligní choroba, autoimunitní či autoinflamatorní choroby, endokarditida, tuberkulóza, ale také MCD) horečka či zvýšená teplota trvá dlouhodobě.
Definice FUO byla zveřejněna v roce 1961, horečku definovali ve stupnici Fahrenheita (101 °F), což převedeno na stupně Celsia činilo 38,3 °C. V roce 2003 Knockaert et al. zjistili, že FUO jen se subfebriliemi a se zvýšenými markery zánětu bývá projevem stejně závažných chorob jako FUO s febriliemi > 38 °C. Ověřili, že FUO se subfebriliemi s ranní tělesnou teplotou ≥ 37,2 °C (≥ 99,0 °F) nebo s tělesnou teplotou naměřenou kdykoliv v průběhu dne ≥ 37,8 °C (≥ 100 °F) provázené zvýšenými hodnotami zánětlivých markerů jsou signálem přítomnosti závažné choroby organizmu. Teplotu měřili v ústech [4,5]. Definici FUO připomínáme v tab. 1 mimo jiné proto, že mezi těmito pacienty FUO je možno nalézt i nemocné s MCD.

V literatuře lze najít více doporučení pro diagnostiku FUO, dle nichž je možné postupovat.
Všechna doporučení pro diferenciální diagnostiku FUO považují FDG-PET/CT zobrazení za velmi přínosné pro diagnostiku FUO. Velká analýza 6 681 pacientů s FUO prokázala, že s pomocí FDG-PET/CT vyšetření byla diagnóza stanovena u 59 % nemocných s FUO [6]. FDG-PET/CT zobrazení může odhalit i obtížně diagnostikovatelné nemoci jako obrovskobuněčnou arteritidu a revmatickou polymyalgii [7].
Následuje biopsie optimální uzliny s vysokou akumulací FDG.V případě klinické suspekce z možné CD by měl být tento údaj spolu s dalšími známými klinickými údaji uveden na žádance k vyšetřované lymfatické uzlině. Patolog v rámci vyšetření lymfatických uzlin u pacientů se známkami systémového zánětu vylučuje kromě infekční etiologie také imunitně/autoimunitně podmíněné léze (vč. CD) a nádorové lymfoproliferace. Upozornění na klinickou suspekci z CD může podpořit i podezření patologa na tuto diagnózu.
Historický vývoj poznání vedl k definici termínů unicentrická CD (unicentric Castleman disease – UCD), která vytváří jedno ložisko, postihuje jednu uzlinu, obvykle bez systémové zánětlivé reakce, a termínu multicentrická CD (multicentric Castleman disease – MCD), která vytváří generalizovanou lymfadenopatii a je pravidelně provázena systémovou zánětlivou reakcí.
S prohlubováním poznání zánětlivých stavů řazených do skupiny CD se postupně vyčleňovaly další formy této choroby.
Členění CD z pohledu roku 2025
Historicky byla jako první popsána UCD a byla pojmenována po autorovi prvního popisu. Teprve později byla popsána MCD, a tak na čas vznikla představa, že existují jen dvě formy, unicentrická, postihující jednu lymfatickou uzlinu, a multicentrická, postihující všechny uzliny. Dle této prvotní představy UCD expandovala pouze lokálně a nebyla provázena systémovou zánětlivou reakcí ani další lymfadenopatií, zatímco MCD byla vždy asociována s generalizovanou lymfadenopatií a systémovými zánětlivými příznaky. To byla původní představa.
Oligocentrická forma CD
A jako u mnoha jiných chorob, tak i zde se postupně zjistilo, že spektrum je kontinuální, a tak na hranici UCD a MCD byla v roce 2021 popsána oligocentrická forma s postižením více uzlin, ale jen v jedné regionální oblasti [8–10], která obvykle nemívá symptomy.
A čínští lékaři u svých zcela asymptomatických pacientů identifikovali lymfadenopatii odpovídající idiopatické CD a popsali soubor asymptomatické CD. Tito pacienti byli dlouhodobě stabilní a nevyžadovali léčbu [11]. Autoři v diskuzi uvádějí, že to mohla být skupina pacientů podobná té, která byla v jiné práci klasifikována jako oligocentrická CD [8–10]. Před akceptováním termínu asymptomatická multicentrická choroba bude třeba jej ověřit v dalších studiích a zjistit, zda tato forma není vázána jen na populaci z Asie.
Unicentrická forma CD (UCD)
UCD obvykle vytváří jedno větší ložisko – nejčastěji v mediastinu, v dutině břišní nebo v pánvi. V < 1/4 případů je ložisko UCD v periferní uzlině. Uzlina s infiltrací UCD bývá na CT či MR zobrazení velmi dobře ohraničená, obvykle je vysoce vaskularizovaná, a proto vykazuje enhancement po podání kontrastu. Někdy jsou v okolí velkého ložiska zřetelné malé satelitní lymfatické uzliny, které po odstranění hlavního ložiska vymizí. Tumorózní zduření tvořené UCD akumuluje FDG středně intenzivně, SUVmax se pohybuje mezi 4 a 6.
Představa, že UCD nevytváří systémové zánětlivé projevy, platí pro 80–90 % případů, zatímco 10–20 % případů je provázeno systémovou zánětlivou reakcí. Na svoji přítomnost upozorní UCD svého nositele bolestmi způsobenými útlakem okolních struktur v důsledku pomalu se zvětšující patologické formace. U některých pacientů je UCD diagnostikována náhodně při zobrazovacím vyšetření indikovaném pro jiný problém.
Unicentrická forma má obvykle hyalinně-vaskulární morfologii (HV) s výrazným zvětšením lymfatické uzliny, proliferací folikulů s regresivně změněnými zárodečnými centry a proliferací cév, často se sklerotizací. V případě MCD není HV morfologie obvykle plně rozvinutá a pro morfologický obraz MCD je upřednostňován termín hypervaskulární změny.
Etiopatogeneze je vysvětlována aktivací stromálních buněk postižené lymfatické uzliny s následnou aktivací B buněk, neovaskularizací, remodelací a postupným zvětšováním ložiska [12].
UCD bývá vzácně asociována s dalšími komplikacemi. Nejčastější z nich je paraneoplastický pemfigus, jehož četnost se v jednotlivých souborech pohybuje mezi 2 a 5 %. Dle literárních popisů může být přítomen před léčbou UCD, ale také se může objevit po odstranění ložiska UCD. Ještě vzácnější komplikací UCD, která však může mít letální následky, je bronchiolitis obliterans. Zcela výjimečnou komplikací může být autoimunita, v literatuře je např. popsána myastenia gravis. U naší poslední pacientky s UCD byla ještě před zjištěním této diagnózy zjištěna roztroušená skleróza.
Z těchto důvodů je doporučováno před operační léčbou UCD pečlivé vyšetření přítomnosti možných mukokutánních ložisek, vyšetření plic případně i s funkčním vyšetřením a laboratorní analýza autoprotilátek obvyklých u pemfigu a u myastenie. Etiopatogenetická souvislost těchto přidružených komplikací není objasněna. Velmi vzácnou komplikací, která byla detekována ve větších souborech řádově v několika procentech, je maligní transformace do sarkomu z folikulárních dendritických buněk (follicular dendritic cell sarcoma), což je onemocnění s velmi nepříznivou prognózou. Léčba UCD je doménou chirurgie [13–15].
Multicentrická CD (MCD)
Později, po popisu UCD, byla rozpoznána MCD, pro niž je charakteristická systémová zánětlivá reakce a generalizovaná lymfadenopatie.
Histopatologicky se rozlišují tři subtypy morfologie – plazmocytární typ, HV, resp. hypervaskulární typ a smíšený typ. Histopatologický obraz změn u MCD se však může vyskytovat i v lymfadenopatiích s jinou zánětlivou či imunitně podmíněnou etiologií nesouvisející s CD, a odlišení může být často problematické. Patolog může konstatovat pouze to, že nález v uzlině spadá do spektra nálezů obvyklých u CD.
Skutečnost je taková, že histopatologický obraz MCD nemá žádný definovaný znak, jehož přítomnost by jasně odlišila zánětlivou lymfadenopatii související s CD od zánětlivé lymfadenopatie nesouvisející s CD. Patolog může pouze konstatovat, že nález v uzlině spadá do spektra nálezů obvyklých u CD.
S postupem poznávání MCD se zjistilo, že není uniformní, a dnes se u ní uvádí i typ oligocentrické formy [8–10]. Etiopatogeneze MCD se považuje za objasněnou jen ve dvou případech:
1. infekce humánním herpesvirem-8 (HHV-8);
2. monoklonální gamapatie, která indukuje jak POEMS syndrom, tak i změny odpovídající CD.
Pro případy, u nichž jsou uvedené příčiny vyloučeny, se používá termínu idiopatická (etiologicky neobjasněná) CD.
MCD s objasněnou etiopatogenezí
Infekce virem Kaposiho sarkomu – HHV-8
Koncem minulého století byla prokázána v uzlinách některých pacientů s MCD infekce HHV-8 neboli virem Kaposiho sarkomu. Pacienti s průkazem HHV-8 byli buď HIV pozitivní, či HIV negativní. A tak byla definována podskupina HHV-8 pozitivní MCD (HHV-8+MCD). U těchto HHV-8+MCD případů je považována infekce HHV-8 virem za etiologický faktor, který vede k nadprodukci interleukinu-6 (IL-6) infikovanými lymfocyty a ten indukuje systémové zánětlivé projevy a lymfadenopatii.
Morfologicky se tato forma někdy vyznačuje proliferací plazmablastů v interfolikulární oblasti (dříve popisovanou jako plasmablastická varianta), která u idiopatické CD není popsána.
Léčba HHV-8 pozitivní CD má za cíl destrukci lymfocytů, v nichž dochází k proliferaci uvedených virů, a má svá specifika v souvislosti s dalšími chorobami, které jsou u HHV-8 pozitivních pacientů přítomny [16–19]. S touto formou choroby nemáme vlastní zkušenosti. Diagnostika HHV-8+MCD vyžaduje vyšetření LANA (latency‐associated nuclear antigen), který se stanoví metodou imunohistochemie v bioptickém vzorku, a vyšetření viremie HHV-8 a HIV ve vzorku periferní krve.
V diferenciální diagnostice HHV-8+MCD je nutno zvažovat HHV-8 pozitivní nehodgkinské lymfomy (buď primary effusion lymphoma, anebo HHV-8 pozitivní difuzní velkobuněčný lymfom s plazmatickou diferenciací). Morfologicky se tato forma někdy vyznačuje plazmablastickou formou, která u idiopatické CD není popsána.
Asociace CD s POEMS syndromem
U některých případů MCD byla zjištěna asociace s POEMS syndromem. POEMS syndrom je klinická jednotka řazená do skupiny monoklonálních gamapatií. Název POEMS je akronym vytvořený z pojmenování charakteristických znaků této nemoci – polyneuropathy, organomegaly, endokrinopathy, monoclonal gammopathy, skin changes. A tak vznikla další podskupina MCD, POEMS-MCD. Etiopatogeneze je v těchto případech spatřována v klonální plazmocelulární populaci a jejích produktech.
Dominantním klinickým projevem POEMS syndromu je polyneuropatie. Viditelnými znaky je novotvorba eruptivních hemangiomů, bělavá barva nehtů a další kožní změny, etiopatogeneticky související s nadprodukcí vaskulárního endoteliálního růstového faktoru (VEGF). Léčba má v těchto případech za cíl eliminaci buněk tvořících monoklonální imunoglobulin [20–23].
Logičnost výše uvedeného členění ale narušují popisy případů POEMS-MCD bez průkazu monoklonálního imunoglobulinu [24]. Někteří autoři uvádějí, že až 15 % případů POEMS syndromu má negativní výsledek imunofixační elektroforézy [25]. To zde trochu zpochybňuje jednotnou etiopatogenezi POEMS-MCD. V těchto případech však nebyla analyzována přítomnost klonálních plazmocytů v kostní dřeni metodou průtokové cytometrie, které je podstatně senzitivnější než mikroskopické vyšetření.
MCD s neobjasněnou etiopatogenezí
Většinu případů MCD tvoří pacienti bez průkazu infekce HHV-8 a bez známek POEMS syndromu. Etiopatogeneze choroby u těchto osob je neznámá, a proto se přidává adjektivum idiopatická (etiologicky neobjasněná). Diagnóza tedy zní idiopatická MCD (iMCD).
Případy spadající do kategorie iMCD se však velmi odlišují intenzitou choroby a závažností poškození, ale i spektrem příznaků a laboratorních nálezů, takže v roce 2025 je tato iMCD rozčleněna do tří podskupin.
Velmi agresivní forma iMCD-TAFRO
Nejagresivnější pól spektra tvoří případy s vysoce intenzivní zánětlivou reakcí spojenou s nekardiální retencí tekutin vedoucí až k anasarce, ale i k poškození funkce ledvin. Trombocytopenie, která iMCD případy provází, může způsobit krvácení. V kostní dřeni těchto pacientů byla detekována retikulinová fibróza.
Velmi agresivní případy iMCD se podobají cytokinové bouři anebo akutnímu průběhu hemofagocytární lymfohistiocytózy [26]. Pro velmi agresivní formy iMCD se dnes používá označení iMCD-TAFRO. Termín TAFRO syndrom je akronym vytvořený z názvů dominujících symptomů (trombocytopenie, anasarka, horečka/retikulinová fibróza, renální dysfunkce, organomegalie). Od méně agresivních forem iMCD ji odlišuje absence výraznějšího vzestupu koncentrace imunoglobulinů. Zvýšení koncentrace polyklonálních imunoglobulinů je typické pro méně agresivní formy iMCD. Také lymfadenopatie nebývá u iMCD-TAFRO nijak výrazná, literatura popisuje průměr postižených uzlin kolem 1,5 mm a tyto uzliny musí vykazovat morfologii odpovídající CD.
První popisy iMCD-TAFRO byly z Asie, ale následně byla iMCD-TAFRO diagnostikována i v populaci Evropy a Severní Ameriky.
iMCD-TAFRO má rychlý nástup, v průběhu 2 měsíců se pacient může dostat do kritického stavu, k lékaři přijde s vysokými horečkami, s intermitentními bolestmi břicha, zácpou, dušností, výrazným zvýšením hmotnosti kvůli retenci tekutin, s velkými edémy. Laboratoř u těchto osob odhalí závažný pokles koncentrace hemoglobinu a trombocytů, velmi vysoké hodnoty CRP a vzestup kreatininu. Imunoglobuliny zde nebývají signifikantně zvýšené. Hodnota feritinu je také zvýšená na hodnoty kolem 950 µg/l, albumin bývá kriticky nízký. V kostní dřeni se popisuje patologický nález – retikulinová fibróza. Histologie odebraných uzlin nachází atrofii zárodečných center s hypervaskularizací, tyto změny může patolog shledat za shodné s histologickým hypervaskulárním obrazem CD [27–33].
Počet popsaných případů iMCD-TAFRO v databázi PubMed se pomalu zvyšuje s tím, jak roste povědomí o této formě [30]. V databázi domácí literatury medvik.cz jsme do října 2025 nenašli popis TAFRO syndromu.
Málo agresivní forma iMCD-IPL
Na opačném, nejméně agresivním pólu spektra iMCD stojí nedávno, po roce 2022, definovaná forma zvaná iMCD-IPL (idiopatická plazmocytární lymfadenopatie).
Tyto případy se vyznačují velmi vysokou koncentrací polyklonálních imunoglobulinů, chronickými zánětlivými projevy, ale jinak příznivou prognózou.
Tato nedávno definovaná forma iMCD-IPL je spojena se zvýšenou četností IgG4 pozitivních plazmocytů ve zvětšených lymfatických uzlinách. Počet IgG4 pozitivních plazmocytů a poměr plazmocytů IgG4+ / plazmocytů IgG+ (IgG4/IgG) se může blížit k hranici 40 %, což je definujícím znakem choroby asociované s imunoglobulinem IgG4 (IgG4-related disease – IgG4-RD). Ne všechny případy iMCD-IPL však tato kritéria splní.
Koncentrace podtřídy imunoglobulinů IgG4 v séru u případů iMCD-ILP bývají zvýšené [34–38]. A tak vlastně tato forma iMCD-IPL je přechodnou klinickou jednotkou mezi IgG4-RD a iMCD.
Zatím nevíme, zda pro iMCD-IPL budou cirkulující plazmablasty v periferní krvi podobně přínosným markerem jako pro IgG4-RD [39].
Středně agresivní forma iMCD-NOS
Mezi iMCD–TAFRO a iMCD–ILP se rozprostírá široké spektrum zbývajících forem, které nejsou ničím dalším specifikované, takže nesou adjektivum not otherwise specified neboli jinak nespecifikované. Domníváme se, že většina našich případů iMCD by spadala do této kategorie. Charakteristiku tří hlavních forem iMCD shrnujeme v tab. 2.
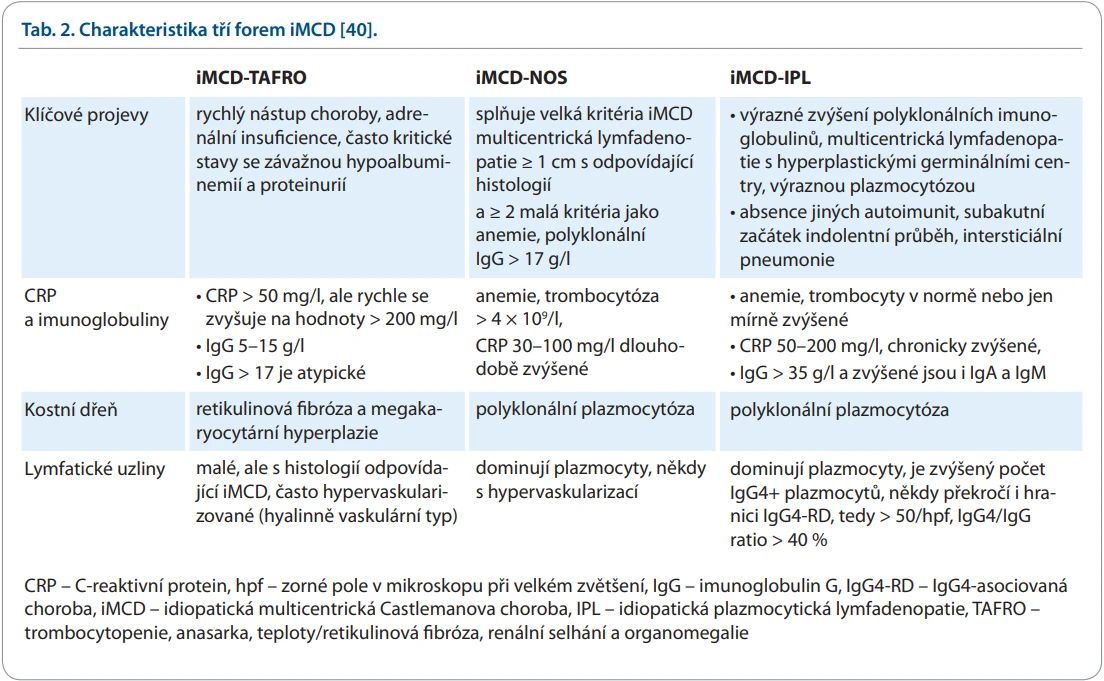
Tito pacienti s blíže nespecifikovanou iMCD (iMCD not otherwise specified – iMCD-NOS) obvykle přicházejí k lékaři se známkami chronického zánětu, hodnoty CRP se pohybují od 50 do 150 mg/l. Jejich lymfatické uzliny jsou mírně zvětšené na průměr 2–3 cm, takže nemusí být klinicky nápadné a bývají rozpoznány až později při zobrazovacím vyšetření. Pacienti s iMCD-NOS mohou mít plicní problémy na podkladě lymfocytární intersticiální pneumonie. Laboratorní nálezy odhalí obvykle mírnou anemii s hemoglobinem kolem 100 g/l, reaktivně zvýšený počet trombocytů a vyšší koncentrace fibrinogenu. Pro tyto případy je typická vysoká hodnota polyklonálních imunoglobulinů, což způsobí i vyšší hodnoty celkové bílkoviny. Koncentrace imunoglobulinů typu IgG se mohou blížit 60 g/l. Histologické vyšetření lymfatických uzlin prokáže nejčastěji polyklonální plazmocytózu těchto uzlin a hyperplastická germinální centra s absencí maligní klonální proliferace [40]. Členění CD z pohledu roku 2025 ilustruje schéma 1, v němž je i orientačně uveden letopočet přijetí uvedené klinické jednotky.
Epidemiologie
CD patří mezi velmi vzácné choroby. Epidemiologické údaje z ČR nemáme k dispozici. Incidence UCD v USA je 16–19 případů na 1 milion obyvatel. Incidence MCD je v USA 5 případů na 1 milion obyvatel [41].
Novější analýza databáze obsahující 30,7 milionu dospělých osob v USA identifikovala mezi lety 2017 a 2018 celkem jen 254 osob s iMCD, čemuž by odpovídala incidence 3,4 na 1 milion dospělých a prevalence 6,9 na 1 milion dospělých [42].
Příznaky iMCD
Choroba má někdy mírné příznaky takzvané flu-like, zatímco v jiných případech může mít závažné příznaky odpovídající cytokinové bouři neboli sepsis-like s anasarkou, s multiorgánovým selháním a smrtí. A k tomu ještě přistupují možné autoimunitní projevy iMCD, takže opravdu každý pacient bude mít odlišný klinický obraz.
Pravidelně se vyskytující projevy zánětu
Mezi pravidelně se vyskytující projevy zánětu patří:
- systémová zánětlivá reakce – subfebrilie, febrilie, noční pocení a úbytek hmotnosti;
- laboratorní nálezy – zvýšené zánětlivé reaktanty jako CRP, feritin a další a s tím spojená anemie, trombocytóza a zvýšená koncentrace fibrinogenu; zánětlivá reakce způsobuje pokles koncentrace albuminu a vzestup koncentrace imunoglobulinů, dominantně imunoglobulinu typu IgG (polyklonální hypergamaglobulinemii), zvyšuje se také plazmatická koncentrace IL-6 a VEGF;
- zobrazovací nálezy – periferní, ale i mediastinální a abdominální lymfadenopatie a hepatosplenomegalie, zvětšení lymfatických uzlin je obvykle jen mírné.
Frekvence uvedených symptomů byla hodnocena v analýze 1 998 pacientů. Laboratorní nálezy byly podobné jak u iMCD, tak u HHV-8 MCD. Zvýšená hodnota CRP byla přítomná u 92,4 %, polyklonální hypergamaglobulinemie u 96,8–100 % nemocných. Anemie provázela 89,4 %, hypoalbuminemie 77 % nemocných. Trombocytopenie byla zaznamenána pouze u 17,3 % osob s iMCD, četnější byla u HHV-8+ případů, kde činila 37,3 %.
V případech UCD se uvedené laboratorní abnormality objevily pouze u 20 % pacientů [43].
Nepravidelně se vyskytující příznaky imunitní etiologie
MCD je často asociována s různými autoimunitními poruchami [43]. Analýza sér 101 pacientů s iMCD identifikovala alespoň jednu reagující autoprotilátku u 47 % vyšetřených osob s iMCD [44]. A v další klinicky orientované práci, analyzující 40 případů iMCD, byly klinické autoimunitní projevy prokázány u 22,5 % pacientů [45]. Autoři těchto analýz uvádějí, že u iMCD dochází velmi často k deregulaci vrozené i získané imunity, a to způsobí, že pacienti s iMCD začnou produkovat autoprotilátky, které dříve netvořili.
Diagnostikující lékař často stojí před otázkou, zda má autoimunitní projevy považovat za kombinaci dvou chorob, nebo za autoimunitní projev iMCD. Mezinárodní doporučení pro diagnostiku uvádí, že kombinace iMCD s další autoimunitní nemocí se má uvádět pouze v případech, kdy jsou kompletně a přesvědčivě naplněna veškerá kritéria autoimunitní choroby. Pokud tomu tak není, jsou jen splněna některá kritéria autoimunitní nemoci, má se na tyto nálezy pohlížet jako na projevy iMCD.
Pokud se v léčbě použije rituximab, je to ale jedno, protože jak autoimunitní choroba, např. Sjögrenův syndrom, tak iMCD lze léčit touto anti-CD20 monoklonální protilátkou [46].
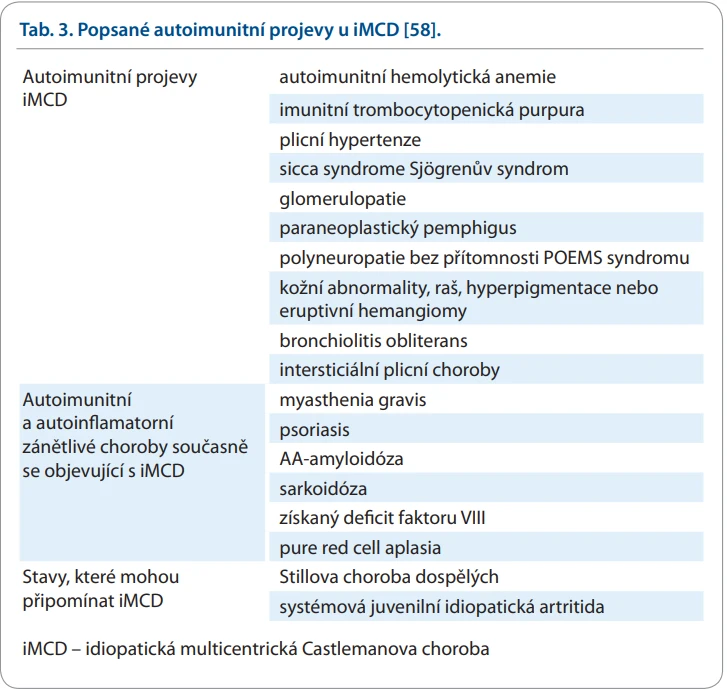
U iMCD je často popisována asociace s následujícími autoimunitními chorobami [47–58]:
- autoimunitní anemie či trombocytopenie, méně často je cytopenie způsobena hemofagocytózou [47–49];
- vaskulitidy, které mohou být příčinou cévní mozkové příhody [50–53];
- paraneoplastický pemphigus [54];
- Sjögrenův syndrom [55];
- myasthenia gravis [54,56];
- kožní změny [57].
V literatuře lze nalézt podstatně více popsaných typů autoimunitních reakcí rozpoznaných u osob s ICD [58]. Přehled popsaných autoimunitních projevů iMCD shrnuje tab. 3.
Nepravidelně se vyskytující příznaky nejasné etiologie
Do této kategorie lze zařadit následující komplikace:
- poškození ledvin endoteliózou v 60 %, AA-amyloidózou ve 20 % – mírné poškození funkce ledvin je popisováno relativně častěji, zatímco závažné poškození funkce ledvin s retencí dusíkatých látek a s anurií je u CD výjimečné;
- nekardiální dušnost a retence tekutin (normální hodnota NTproBNP) – ascites, plicní výpotek, plicní abnormality typu infiltrátů, restriktivní plicní poruchy, intersticiální pneumonie, či dokonce bronchiolitis obliterans;
- kožní změny vč. hyperpigmentace, kožní raš, tvorba kožních hemangiomů [57];
- zcela výjimečně průjmy a hmotností úbytek;
- dysfunkce jater [43,59].
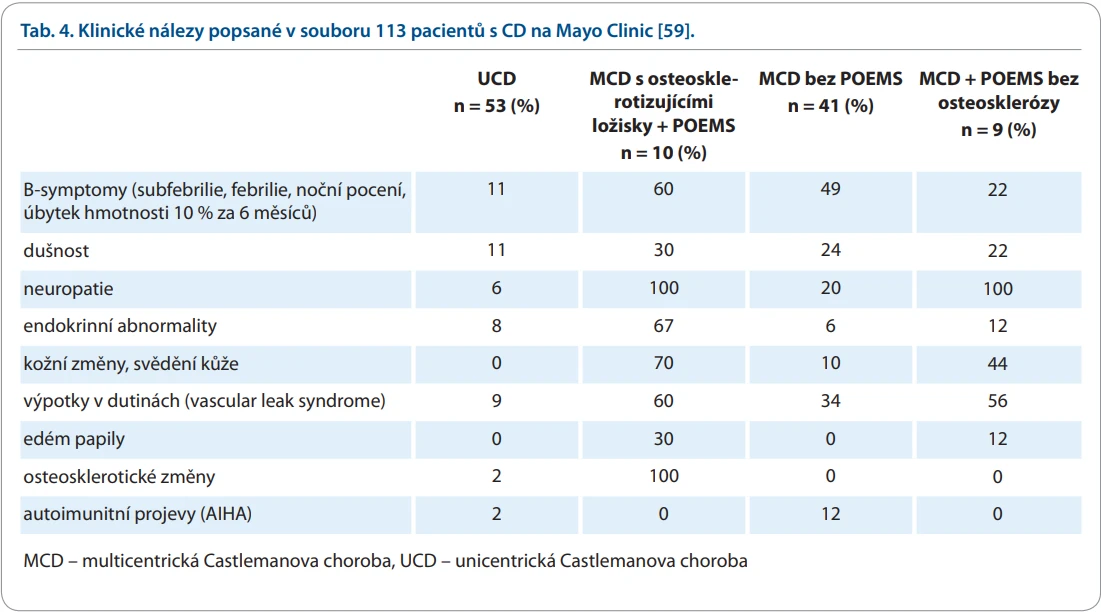
Uvedený výčet možných komplikací je opravdu široký a vysvětluje, proč je stanovení diagnózy tak obtížné. Pestrost projevů CD doložíme ještě přehledem symptomů a projevů, jak byl zaznamenán na souboru z Mayo Clinic v USA [59], v tab. 4.
Stanovení diagnózy
Stanovení diagnózy UCD je obvykle následkem chirurgického výkonu s následným histologickým hodnocením materiálu. Unicentrická forma má ve většině případů HV strukturu, která nečiní problémy při histologickém stanovení diagnózy.
Stanovení diagnózy iMCD závisí na spolupráci patologa a klinika. Patolog musí od klinika dostat informaci, že by se mimo jiné mohlo jednat o iMCD. A pak se patolog na vyšetřovaný preparát musí dívat z úhlu pohledu informací, které přináší doporučení pro histopatologickou diagnostiku této nemoci [60–62].
Pokud patolog informace o možné diagnóze iMCD nemá, může naopak být, při splnění morfologických kritérií, vč. adekvátního gradingu histomorfologických změn, iniciátorem klinického dovyšetření pacienta s přihlédnutím k platným kritériím.
Pokud patolog uvede, že histomorfologický obraz by mohl mít vysvětlen v CD, tak na to naváže klinik srovnáním nálezů konkrétního pacienta s recentními kritérii této choroby.
První mezinárodní kritéria iMCD formulovali Faigenbaum et al. v roce 2017 (tab. 5) [60] a na to navázalo doporučení pro léčbu [63].

V roce 2021 byla přijata validovaná kritéria iMCD-TAFRO, která uvádí tab. 6 [64]. Předtím ale již více let existoval návrh iMCD-TAFRO kritérií.

Nedávno byla nově koncipována jednotka iMCD-IPL, byl zveřejněn její popis, byť zatím nebyla definována kritéria této choroby [34–38].
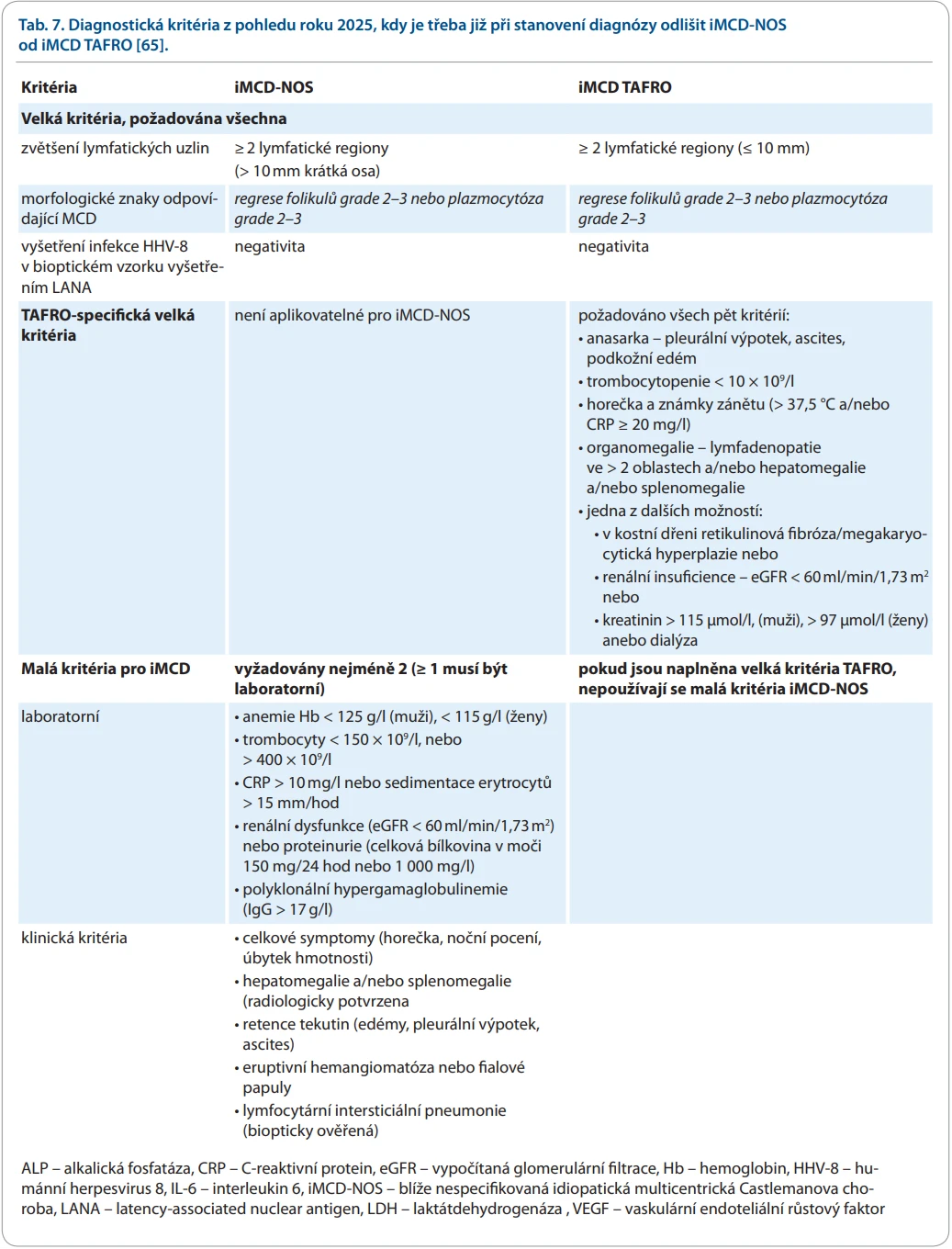
Tento vývoj se odrazil v publikaci Alnoora et al. [65], kteří v roce 2025 nastavili vývoji kritérií zrcadlo a sumarizovali současnou klasifikaci idiopatické CD do skupiny iMCD-NOS a iMCD-TAFRO. Autoři tab. 7 považují za žádoucí ihned zpočátku oddělit tyto skupiny, protože iMCD-TAFRO vyžaduje intenzivnější léčbu. V této verzi kritérií iMCD-NOS již není hypoalbuminemie uvedena jako malé kritérium, jak bylo v původní verzi z roku 2017.

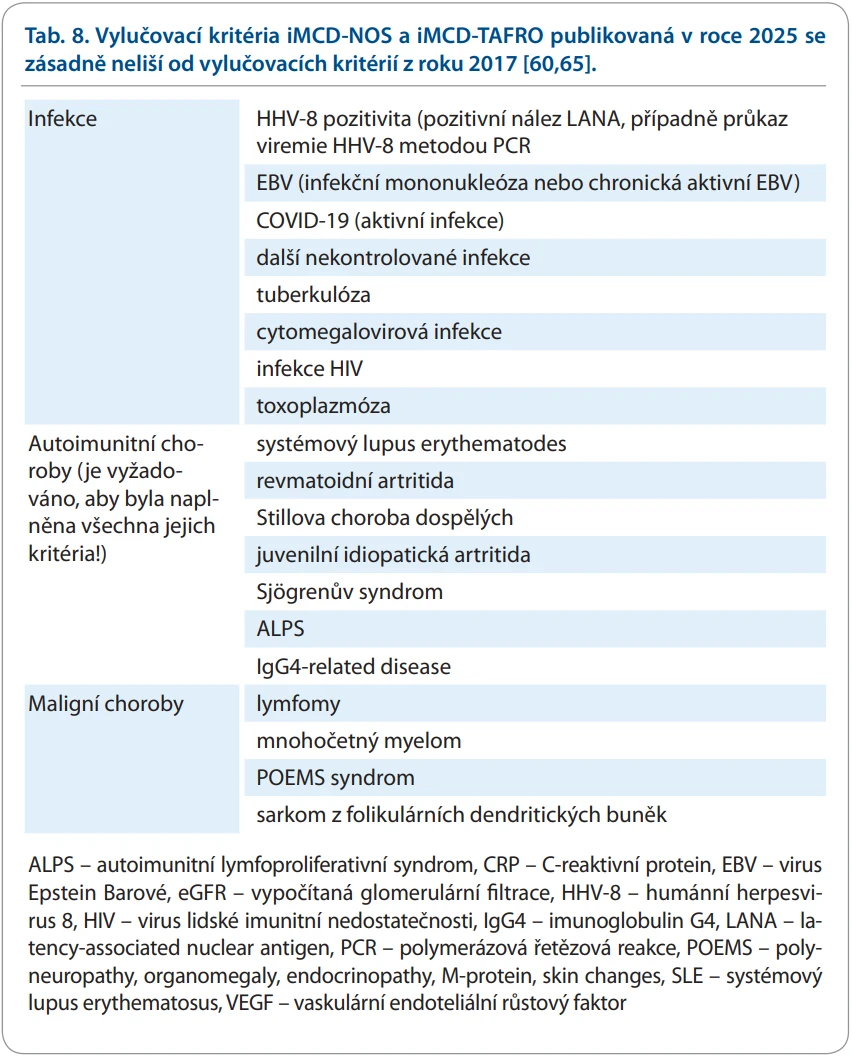
Pokud jsou uvedená kritéria naplněna, je silné podezření na tuto chorobu. Pro definitivní diagnózu je třeba ještě vyloučit jiné nemoci, které mohou indukovat podobný morfologický obraz. Vylučovací kritéria z roku 2017 a 2025 se nijak neliší a jsou uvedena v tab. 8 [60,65].
Život ohrožující iMCD-TAFRO je málo častá forma. Snad proto je spojena s otázkami. Pokud lymfadenopatie splní kritéria iMCD a ostatní příznaky kritéria TAFRO, tak se jedná o iMCD-TAFRO. Jsou ale popsány i případy bez nálezu známek CD v odebrané uzlině, pak by se jednalo jen o TAFRO syndrom [66].
Přínos pozitronové emisní tomografie
V dnešní době je považováno za optimální zobrazení s aplikací FDG, tedy FDG-PET/CT případně FDG-PET/MR. Pro iMCD je typická střední akumulace FDG hodnoty SUVmax pohybující se mezi 4 a 6, výjimečná je vysoká akumulace s SUVmax 10 [67,68]. Podrobně je FDG-PET/CT obraz 29 pacientů analyzován v práci autorů z Masarykova onkologického ústavu v Brně [69].
Diagnostika plicních projevů CD
Z uvedeného výčtu projevů CD je zřejmé, že pro přesnou diagnostiku těchto komplikací je třeba specialistů na dané oblasti medicíny. Jak UCD, tak MCD je vzácně spjata s plicními projevy typu bronchiolis obliterans a s lymfocytární pneumonií, jejich diagnostika spadá do kompetence specialistů na plicní choroby.
Bronchiolitis obliterans se může vyskytovat u CD v důsledku chronické zánětlivé aktivity s nadprodukcí cytokinů (zejména IL-6), která vede k poškození malých dýchacích cest, fibrózní remodelaci bronchiolů a následně obstrukci. Diagnostika bronchiolitis obliterans u pacientů s CD je založena na kombinaci klinického obrazu (progredující dušnost, suchý kašel, poslechově pískoty), plicním funkčním vyšetření (obstrukční ventilační porucha, hodnota FEV1 – usilovně vydechnutý objem za 1 s, poměr FEV1/FVC – forsírovaná vitální kapacita) a HRCT hrudníku (nález mozaikové perfuze, air-trappingu a bronchiolárních změn). U pacientů splňujících klinická, funkční a CT kritéria není bronchoskopie vyžadována. Bronchoskopie s bronchoalveolární laváží je prováděna za účelem vyloučení infekční příčiny. Transbronchiální biopsie / kryobiopsie je indikována individuálně při nejednoznačném nálezu.
Lymfoidní intersticiální pneumonie je vzácná intersticiální plicní choroba, typicky asociovaná s poruchami imunity, jako je Sjögrenův syndrom, systémový lupus erythematodes, a mimo jiné je také popisována u CD a je i v kritériích této nemoci. Její průběh je subakutní až chronický s nespecifickými příznaky, jako jsou kašel a dušnost. Může způsobovat cystické projevy podobně jako histiocytóza z Langerhansových buněk. Nespecifické radiologické nálezy jsou důvodem, proč definitivní diagnózu lze stanovit pouze biopsií [70–73].
Diferenciální diagnóza
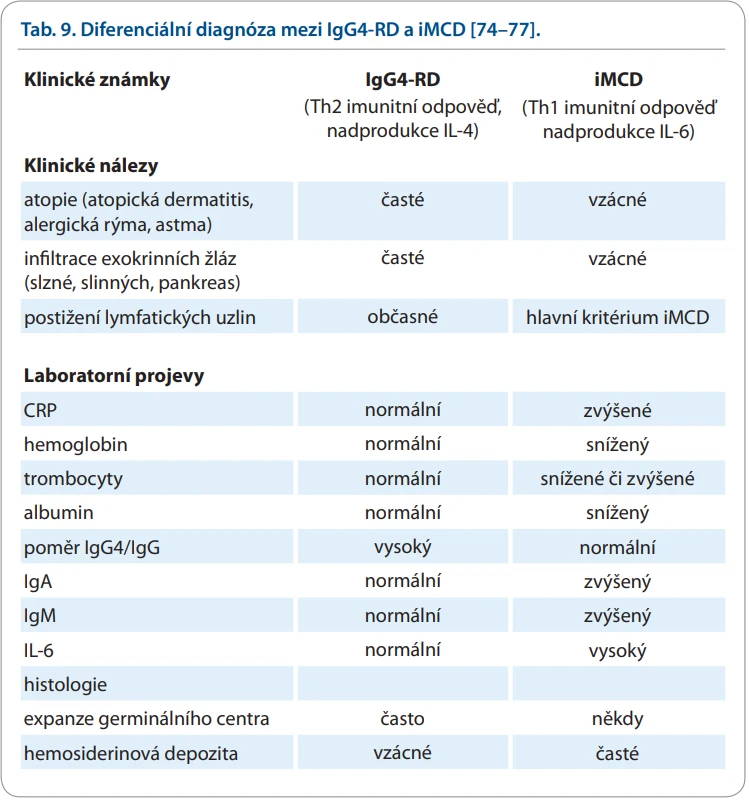
Podezření na iMCD vzniká u pacientů s polyklonální hypergamaglobulinemií. Ta může mít více příčin [74]. Zde uvedeme výsledky kanadské analýzy, která je nejpodrobnější. Má ale vyšší počet diagnóz IgG4-RD, než obsahuje analýza z Mayo Clinic.
Autoři analyzovali pacienty vyšetřené v nemocnici Vancouver General Hospital od října 2016 až do listopadu 2017 [75]. Zjištěné diagnózy byly rozřazeny celkem do šesti kategorií, sedmá kategorie byla vyhrazena pro jiné nemoci, nezařazené do předchozích kategorií:
1. imunitní zánětlivé nemoci vč. systémových nemocí pojiva a iMCD 31,8 % (21 z 66);
2. krevní choroby vč. maligních 21,2 % (14 z 66);
3. IgG4-RD 19,6 % (13 z 66);
4. nemoci jater vč. hepatitid 10,6 % (7 z 66);
5. infekční nemoci vyjma hepatitid 4,5 % (3 z 66);
6. nehematologické malignity 1,5 % (1 z 66);
7. jiné příčiny, nespadající do uvedených kategorií 10,6 % (7 z 66) [75].
Citované práce jsou dobrým pomocníkem pro diferenciální diagnostiku polyklonální hypergamaglobulinemie [74–77].
Z obrovské skupiny imunitních zánětlivých chorob upozorníme na tři, které mají následující společnou charakteristiku:
- výrazná polyklonální hypergamaglobulinemie, a tím i hyperproteinemie;
- zánětlivá infiltrace lymfatických uzlin, exokrinních žláz a případně dalších orgánů;
- absence jednoznačného diagnostického znaku, a tedy stanovování diagnóz dle mezinárodních kritérií.
Jedná se o iMCD, dále pak IgG4-RD a Sjögrenův syndrom. Společnou etiopatogenezí všech těchto chorob je alterace imunity vedoucí k expanzi B buněk, k jejich diferenciaci v plazmocyty a ke tvorbě polyklonálních imunoglobulinů typu IgG a k zánětlivé reakci, která však u IgG4-RD a u iMCD má odlišné mediátory.
V literatuře lze nalézt hodně prací, které se snaží hledat hranice mezi těmito třemi chorobami [78–83]. Doporučení k odlišení těchto dvou chorob shrnuje tab. 9.
Průběh iMCD
iMCD je typicky diagnostikována ve 4. a 5. dekádě života, častěji u mužů než u žen.
Ve většině případů má choroba dlouholetý průběh, pacientům sice nebere rychle život, ale výrazně zhoršuje kvalitu jejich života. Projevy případné vaskulitidy mohou trvale invalidizovat (cévní mozkové příhody) a komplikují další léčbu i ošetřování. Vzhledem k narušení imunity se zvyšuje u těchto nemocných výskyt dalších maligních chorob ve srovnání s průměrnou populací. Literatura popisuje následující čtyři formy klinického průběhu:
- opakované relapsy a remise;
- stabilní perzistující choroba;
- progredující fatální choroba (sepsis-like);
- transformace v maligní lymfom.
Mezinárodní kritéria pro léčbu rozdělují léčebné postupy při flu-like průběhu a sepsis-like průběhu a definují tyto stavy.
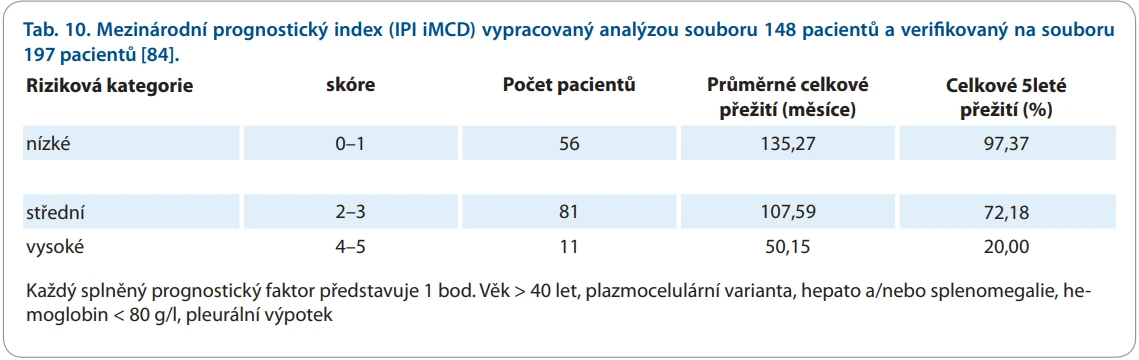
Prognóza choroby souvisí se závažností klinických projevů, jak vyplývá z mezinárodního prognostického indexu (tab. 10) [84]. Tento index potvrzuje značnou diverzitu závažnosti choroby.
Závěr
V textu jsme stručně charakterizovali CD z pohledu roku 2025. Oproti situaci před 5 lety zde máme několik nových podskupin této choroby, vč. nových kritérií TAFRO syndromu. Cílem textu je přispět k časnější diagnostice těchto chorob.
Domníváme se, že počet diagnostikovaných případů souvisí jednoznačně s tím, jak často si lékaři vyšetřující zánětlivé a autoimunitní stavy na tuto diagnózu vzpomenou a analyzují příznaky a nálezy pacienty podle kritérií této choroby. Může být příčinou i retroperitonální masy imitující retroperitonální fibrózu [85].
Velkým kritériem je histomorfologický obraz a zde je nutno se cíleně obrátit na patologa s otázkou, zda nález v lymfatické uzlině může být kompatibilní s touto diagnózou. Patolog může konstatovat, že nález může odpovídat této nemoci, nikoliv že se o tuto diagnózu jednoznačně jedná, resp. že je nutné doplnění klinické korelace. Diagnóza iMCD je tedy klinicko-patologická, to znamená, že je výsledkem diskuze mezi patologem a klinikem.
Léčbě této nemoci věnujeme samostatný text. Jak UCD, tak iMCD patří sice mezi vzácnější choroby, ale přesto se lékaři u nás s touto chorobou opakovaně setkávali, mnohdy byli překvapeni různorodými projevy u konkrétních pacientů, ale přesto se jim ji podařilo diagnostikovat a léčit. Tyto zajímavé domácí publikace citujeme až v závěru textu, abychom ilustrovali různorodé projevy této choroby a doložili, že u nás je mnoho lékařů, kteří mají s diagnostikou a léčbou této choroby zkušenosti, a je možné se tedy na ně obrátit s žádostí o konzultaci problému, který je podobný tomu, čemu se ve svých publikacích věnovali [86–98].
Léčba CD je podrobně popsána v článku, který vyšel v předchozím čísle Klinické onkologie [99].
Dedikace
Podpořeno: MZ ČR – RVO (FNBr, 65269705 a MOÚ, 00209805).
Zdroje
1. Adam Z, Mayer J, Pour L et al. Autoinflamatorní choroby se symptomy, které připomínají maligní krevní choroby – syndrom Schnitzlerové, Stillova choroba dospělých, SAPHO a VEXAS syndrom. Transfuze Hematol dnes 2024; 30 (3): 151–168. doi: 10.48095/cctahd2024prolekare.cz13.
2. Alaggio R, Amador C, Anagnostopoulos I et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia 2022; 36 (7): 1720–1748. doi: 10.1038/s41375-022-01620-2.
3. Schmalzing M, Sander O, Seidl M et al. Castleman’s disease in the rheumatological practice. Z Rheumatol 2024; 83 (Suppl 3): 289–298. doi: 10.1007/s00393-024-01560-5.
4. Petersdorf RG, Beeson PB. Fever of unexplained origin: report on 100 cases. Medicine (Baltimore) 1961; 40 : 1–30. doi: 10.1097/00005792-196102000-00001.
5. Knockaert DC, Vanderschueren S, Blockmans D. Fever of unknown origin in adults: 40 years on. J Intern Med 2003; 253 (3): 263–275. doi: 10.1046/j.1365-2796.2003.01120.x.
6. Romano Gargarella E, Vocaturo F, Guarneri A et al. Role of 18F FDG-PET-CT in fever and inflammation of unknown origin. J Clin Med 2025; 14 (16): 5861. doi: 10.3390/jcm14165861.
7. Řehák Z, Šprláková-Puková A, Kazda T et al. 18F-FDG PET/CT in polymyalgia rheumatica-a pictorial review. Br J Radiol 2017; 90 (1076): 20170198. doi: 10.1259/bjr.20170198.
8. Pierson SK, Bagg A, Alapat D et al. Characterization of Castleman disease reveals patients with oligocentric adenopathy and clinicopathologic characteristics similar to unicentric Castleman disease. Blood 2021; 138 (Suppl 1): 1622. doi: 10.1182/blood-2021-153840.
9. Pierson SK, Brandstadter JD, Torigian DA et al. Characterizing the heterogeneity of castleman disease and oligocentric subtype: findings from the ACCELERATE registry. Blood Adv 2025; 9 (8): 1952–1965. doi: 10.1182/bloodadvances.2024014391.
10. Beckham TH, Yang JC, Chau KW et al. Excellent outcomes with surgery or radiotherapy in the management of castleman disease including a case of oligocentric disease. Clin Lymphoma Myeloma Leuk 2020; 20 (10): 685–689. doi: 10.1016/j.clml.2020.05.002.
11. Zhang L, Liu QH, Zhou H et al. Asymptomatic multicentric castleman disease: a potential early stage of idiopathic MCD. Blood Adv 2024; 8 (21): 5598–5602. doi: 10.1182/bloodadvances.2024013728.
12. Smith D, Eichinger A, Fennell É et al. Spatial and single cell mapping of Castleman disease reveals key stromal cell types and cytokine pathways. Nat Commun 2025; 16 (1): 6009. doi: 10.1038/s41467-025-61214-1.
13. van Rhee F, Oksenhendler E, Srkalovic G et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv 2020; 4 (23): 6039–6050. doi: 10.1182/bloodadvances.2020003334.
14. Horváth T, Frola L, Adam Z et al. Léčba unicentrické Castlemanovy choroby rituximabem, bendamustinem a dexametazonem zmenšila objem expanzivního ložiska v horním mediastinu a umožnila jeho radikální odstranění. Klin Onkol 2025; 38 (2): 132–143. doi: 10.48095/ccko2025132.
15. Penka I, Kala Z, Zetelová A. et al. Casteman disease – surgical treatment, case reports. Rozhl Chir 2016; 95 (12): 457–461.
16. Dunn R, Jariwal R, Venter F et al. HHV-8-associated multicentric Castleman disease, a diagnostic challenge in a patient with acquired immunodeficiency syndrome and fever. J Investig Med High Impact Case Rep 2022; 10 : 23247096221097526. doi: 10.1177/23247096221097526.
17. Osa M, Maeda T, Misawa K et al. Clinical response to liposomal doxorubicin and rituximab in HHV-8-associated multicentric Castleman’s disease in an HIV-positive patient. J Infect Chemother 2016; 22 (12): 804–807. doi: 10.1016/j.jiac.2016.06.004.
18. Kaga H, Kurahashi H, Kubota A et al. Successful rituximab treatment of an elderly Japanese patient with HHV8--positive, HIV-negative multicentric castleman disease. Int J Hematol 2022; 115 (1): 129–134. doi: 10.1007/s12185-021-03222-7.
19. Murphy C, Hawkes E, Chionh F et al. Durable remission of both multicentric Castleman’s disease and Kaposi’s sarcoma with valganciclovir, rituximab and liposomal doxorubicin in an HHV-8-positive, HIV-negative patient. J Clin Pharm Ther 2017; 42 (1): 111–114. doi: 10.1111/jcpt.12472.
20. Min C, Liu A, Xu Y et al. A case report of Castleman disease variant of POEMS syndrome presenting with prominent polyserositis and renal impairment. Front Med (Lausanne) 2025; 12 : 1537944. doi: 10.3389/fmed.2025.1537944.
21. Brioli A, Wyrwa A, Rüddel U et al. Mutations in the plasma cell clone identify mechanism of polyneuropathy in a case of POEMS syndrome associated with Castleman disease and multiple myeloma. Ann Hematol 2023; 102 (1): 239–242. doi: 10.1007/s00277-022-05032-1.
22. Lee YM, Choi YS, Kim JM. POEMS syndrome: presented as idiopathic multicentric Castleman disease of plasma cell variant for eight years and dramatic treatment with siltuximab followed by autologous peripheral blood stem cell transplantation. Diagnostics (Basel) 2022; 12 (4): 998. doi: 10.3390/diagnostics12040998.
23. Minařík J, Ščudla V, Bačovský J et al. POEMS syndrom. Onkologie (Olomouc) 2011; 5 (3): 151–154.
24. Ji M, Jin S, Zang S et al. Castleman disease variant of POEMS syndrome without M protein: a case report. Front Oncol 2024; 14 : 1449945. doi: 10.3389/fonc.2024.1449945.
25. Prokop J, Estorninho J, Marote S et al. POEMS syndrome: a rare cause of adrenal insufficiency in a young male. Endocrinol Diabetes Metab Case Rep 2019; 2019: EDM190010. doi: 10.1530/EDM-19-0010.
26. Fajgenbaum DC. The cytokine storm of multicentric Castleman disease. Adv Exp Med Biol 2024; 1448 : 459–467. doi: 10.1007/978-3-031-59815-9_31.
27. Zhou QY. Castleman disease and TAFRO syndrome: to improve the diagnostic consciousness is the key. World J Clin Cases 2022; 10 (5): 1536–1547. doi: 10.12998/wjcc.v10.i5.1536.
28. Godfrey K, Harris E, Moss H et al. Idiopathic multicentric castleman disease of TAFRO subtype. Br J Haematol 2022; 196 (3): 461. doi: 10.1111/bjh.17843.
29. Takai K. TAFRO syndrome: a syndrome or a subtype of multicentric Castleman disease? Biomedicines 2024; 12 (3): 652. doi: 10.3390/biomedicines12030652.
30. Nishimura Y, Nishimura MF, Fajgenbaum DC et al. Global public awareness of Castleman disease and TAFRO syndrome between 2015 and 2021: a google trends analysis. EJHaem 2022; 3 (3): 748–753. doi: 10.1002/jha2.459.
31. Lossos C, Brown J, Sheikhbahaei S et al. Idiopathic multicentric Castleman disease – TAFRO results in high levels of mtor activator SVEP1, tissue factor, and endotheliopathy. Blood Vessel Thromb Hemost 2024; 1 (2): 100006. doi: 10.1016/j.bvth.2024.100006.
32. Lust H, Gong S, Remiker A et al. Idiopathic multicentric Castleman disease with TAFRO clinical subtype responsive to IL-6/JAK inhibition: a pediatric case series. Pediatr Blood Cancer 2021; 68 (10): e29261. doi: 10.1002/pbc.29261.
33. Nishimura Y, Hanayama Y, Fujii N et al. Comparison of the clinical characteristics of TAFRO syndrome and idiopathic multicentric Castleman disease in general internal medicine: a 6-year retrospective study. Intern Med J 2020; 50 (2): 184–191. doi: 10.1111/imj.14404.
34. Gao YH, Liu YT, Zhang MY et al. Idiopathic multicentric Castleman disease (iMCD) -idiopathic plasmacytic lymphadenopathy: a distinct subtype of iMCD-not otherwise specified with different clinical features and better survival. Br J Haematol 2024; 204 (5): 1830–1837. doi: 10.1111/bjh.19334.
35. Nishikori A, Nishimura MF, Fajgenbaum DC et al. Diagnostic challenges of the idiopathic plasmacytic lymphadenopathy (IPL) subtype of idiopathic multicentric Castleman disease (iMCD): factors to differentiate from IgG4-related disease. J Clin Pathol 2025; 79 (1): 43–49. doi: 10.1136/jcp-2023-209280.
36. Nishikori A, Nishimura MF, Nishimura Y et al. Idiopathic plasmacytic lymphadenopathy forms an independent subtype of idiopathic multicentric Castleman disease. Int J Mol Sci 2022; 23 (18): 10301. doi: 10.3390/ijms231810301.
37. Yen CC, Chen TY. Idiopathic plasmacytic lymphadenopathy with polyclonal hypergammaglobinemia mimicking plasma cell myeloma. Blood 2018; 132 (25): 2700. doi: 10.1182/blood-2018-07-863290.
38. Kojima M, Nakamura N, Otuski Y et al. Pulmonary lesion of idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia appears to be a cause of lymphoplasmacytic proliferation of the lung: a report of five cases. Pathol Res Pract 2008; 204 (3): 185–190. doi: 10.1016/j.prp.2007.11.003.
39. Tartakover Matalon S, Rabinowicz N, Carmi O et al. Examination of the level of circulating plasmablasts and their characteristics as diagnostic tools for immunoglobulin G4-related disease. Isr Med Assoc J 2024; 26 (6): 369–375.
40. Chen LYC, Zhang L, Fajgenbaum DC. Expert perspective: diagnosis and treatment of Castleman disease. Arthritis Rheumatol 2026; 78 (1): 12–25. doi: 10.1002/art.43269.
41. Munshi N, Mehra M, van de Velde H et al. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma 2015; 56 (5): 1252–1260. doi: 10.3109/10428194.2014.953145.
42. Mukherjee S, Martin R, Sande B et al. Epidemiology and treatment patterns of idiopathic multicentric castleman disease in the era of IL-6-directed therapy. Blood Adv 2022; 6 (2): 359–367. doi: 10.1182/bloodadvances.2021004441.
43. Hoffmann C, Oksenhendler E, Littler S et al. The clinical picture of Castleman disease: a systematic review and meta-analysis. Blood Adv 2024; 8 (18): 4924–4935. doi: 10.1182/bloodadvances.2024013548.
44. Feng A, Gonzalez MV, Kalaycioglu M et al. Common connective tissue disorder and anti-cytokine autoantibodies are enriched in idiopathic multicentric Castleman disease patients. Front Immunol 2025; 16 : 1528465. doi: 10.3389/fimmu.2025.1528465.
45. Nakazato Y, Tsuchida S, Takada-Owada A et al. Castleman disease and mimickers: clinicopathological findings of atypical lymphoproliferative disorders associated with autoimmune disease. J Clin Exp Hematop 2022; 62 (3): 119–126. doi: 10.3960/jslrt.22025.
46. Kaegi C, Wuest B, Schreiner J et al. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol 2019; 10 : 1990. doi: 10.3389/fimmu.2019.01990.
47. Cáceres Medina JL, González Torres LA, Gamboa--Meza A et al. Rituximab as a first-line treatment for autoimmune hemolytic anemia in multicentric Castleman’s disease. Cureus 2024; 16 (4): e59080. doi: 10.7759/cureus.59080.
48. Iskandar A, Hwang A, Dasanu CA. Severe warm-antibody autoimmune hemolytic anemia due to multicentric castleman disease: responding to rituximab. J Oncol Pharm Pract 2019; 25 (8): 2016–2018. doi: 10.1177/1078155218816775.
49. Plano F, Mancuso S, Camarda GM et al. A multicentric Castleman disease associated with mixed warm and cold antibody-mediated AHA responsive to siltuximab. Chemotherapy 2024; 69 (1): 35–39. doi: 10.1159/000533235.
50. Qian Y, Zhang M. Retinal vasculitis associated with Castleman disease. JAMA Ophthalmol 2025; 143 (1): e245152. doi: 10.1001/jamaophthalmol.2024.5152.
51. Rössler M, Kiessling B, Klotz JM et al. Recurrent cerebral ischemias due to cerebral vasculitis within the framework of incomplete POEMS syndrome with Castleman disease. Nervenarzt 2004; 75 (8): 790–794. doi: 10.1007/s00115-004-1683-x.
52. Yin H, Zhang L, Pan B et al. Cerebral thrombotic event as a rare complication in a young female patient with idiopathic multicentric Castleman disease. Clin Neurol Neurosurg 2020; 198 : 106246. doi: 10.1016/j.clineuro.2020.106246.
53. Goda T, Oyama N, Iwamoto T et al. A case of cerebral infarction in a patient with TAFRO syndrome. J Neurol Sci 2019; 400 : 21–22. doi: 10.1016/j.jns.2019.03.006.
54. Jakubíková M, Piťha J, Latta J et al. Myasthenia gravis, Castleman disease, pemphigus, and anti-phospholipid syndrome. Muscle Nerve 2013; 47 (3): 447–451. doi: 10.1002/mus.23657.
55. Pan Y, Cui Z, Wang S et al. Idiopathic multicentric Castleman disease with Sjögren’s syndrome and secondary membranous nephropathy: a case report and review of the literature. BMC Nephrol 2020; 21 (1): 528. doi: 10.1186/s12882-020-02191-z.
56. Nandy S, Bhattacharya D, Kundu S et al. Myasthenia gravis with Castleman disease: a case report with review of literature. Indian J Pathol Microbiol 2025; 68 (2): 392–394. doi: 10.4103/ijpm.ijpm_482_23.
57. Kim HJ, Han JH, Bang CH et al. Cutaneous disorders associated with Castleman’s disease. Acta Derm Venereol 2019; 99 (11): 984–989. doi: 10.2340/00015555-3253.
58. González García A, Fernández-Martín J, Robles Marhuenda Á. Idiopathic multicentric castleman disease and associated autoimmune and autoinflammatory conditions: practical guidance for diagnosis. Rheumatology (Oxford) 2023; 62 (4): 1426–1435. doi: 10.1093/rheumatology/keac481.
59. Dispenzieri A, Armitage JO, Loe MJ. et al. The clinical spectrum of Castleman’s disease. Amer J Hematol 2012; 87 (11): 997–1002. doi: 10.1002/ajh.23291.
60. Fajgenbaum DC, Uldrick TS, Bagg A et al. International, evidence-based consensus diagnostic criteria for HHV--8-negative/idiopathic multicentric Castleman disease. Blood 2017; 129 (12): 1646–1657. doi: 10.1182/blood-2016-10-746933.
61. Gasljevic G, Bonometti A, Anagnostopoulos I et al. The morphological spectrum of Castleman disease and related disorders: a report from the lymphoma workshop of the 22nd meeting of the European Association of Hematopathology. Virchows Arch 2025; 487 (2): 253–273. doi: 10.1007/s00428-025-04171-w.
62. Nishimura MF, Haratake T, Nishimura Y et al. International consensus histopathological criteria for subtyping idiopathic multicentric Castleman disease based on machine learning analysis. Am J Hematol 2025; 100 (9): 1502–1512. doi: 10.1002/ajh.27743.
63. van Rhee F, Voorhees P, Dispenzieri A et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018; 132 (20): 2115–2124. doi: 10.1182/blood-2018-07-862334.
64. Nishimura Y, Fajgenbaum DC, Pierson SK et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol 2021; 96 (10): 1241–1252. doi: 10.1002/ajh.26292.
65. Alnoor F, Spies NC, Kumar J et al. The evolution and recent advances in diagnostic criteria for idiopathic multicentric Castleman disease. Am J Hematol 2025; 100 (11): 2064–2073. doi: 10.1002/ajh.70039.
66. Li SY, Gao YH, Dang Y et al. TAFRO Syndrome without pathology supporting Castleman disease: to be treated as idiopathic multicentric Castleman disease-tafro or a distinct disease entity? Turk J Haematol 2025; 42 (1): 1–8. doi: 10.4274/tjh.galenos.2025.2024.0420.
67. Wang G, Xu Q, Liu Y et al. 18F-FDG PET/CT Metabolic parameters are correlated with clinical features and valuable in clinical stratification management in patients of Castleman disease. Cancer Imaging 2025; 25 (1): 12. doi: 10.1186/s40644-025-00833-9.
68. Iguchi T, Nishikori A, Sato Y et al. Computed tomography findings of idiopathic multicentric Castleman disease subtypes. J Clin Exp Hematop 2024; 64 (4): 292–296. doi: 10.3960/jslrt.24053.
69. Koukalová R, Selingerová I, Řehák Z et al. FDF-PET/CT v diagnostice a hodnocení léčebné odpovědi, retrospektivní studie 29 případů z jednoho centra. Klin Onkol 2021; 34 (2): 120–127. doi: 10.48095/ccko2021120.
70. Cui Z, Zhou X, Luo F et al. Worldwide bronchiolitis obliterans research: a bibliometric analysis of the published literature between 2002 and 2022. Medicine (Baltimore) 2023; 102 (28): e34263. doi: 10.1097/MD.0000000000034263.
71. Bartková H, Válková V, Vydra J et al. Bronchiolitis obliterans syndrom jako projev chronické reakce štěpu proti hostiteli po allogenní transplantaci krvetvorných buněk. Stud Pneumol Phthiseol 2021; 81 (3): 112–120.
72. Lau HX, Krebs L, Sim S et al. Lymphoid interstitial pneumonia without known cause: diagnostic work up and differential considerations. Respirol Case Rep 2025; 13 (4): e70175. doi: 10.1002/rcr2.70175.
73. Doubková M, Doubek M, Richter S et al. Běžná variabilní imunodeficience a granulomatózní/lymfocytární intersticiální plicní nemoc (GLILD). Stud Pneumol Phthiseol 2023; 83 (5): 159–169.
74. Dispenzieri A, Gertz MA, Therneau TM et al. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76 (5): 476–487. doi: 10.4065/76.5.476.
75. Zhao EJ, Carruthers MN, Li CH et al. Conditions associated with polyclonal hypergammaglobulinemia in the IgG4-related disease era: a retrospective study from a hematology tertiary care center. Haematologica 2020; 105 (3): e121–e123. doi: 10.3324/haematol.2019.219725.
76. Andre M, Contis A, Berard AM. Etiological study of polyclonal hypergammaglobulinemia in a French cohort of hospitalized patients and proposal of a diagnostic aid algorithm: short title: polyclonal hypergammaglobulinemia in a French cohort of hospitalized patients. Sci Rep 2024; 14 (1): 31282. doi: 10.1038/s41598-024-82735-7.
77. Beuvon C, Martin M, Baillou C et al. Etiologies of polyclonal hypergammaglobulinemia: a scoping review. Eur J Intern Med 2021; 90 : 119–121. doi: 10.1016/j.ejim.2021.05.023.
78. Sasaki T, Akiyama M, Kaneko Y et al. Immunoglobulin G4-related disease and idiopathic multicentric Castleman’s disease: confusable immune-mediated disorders. Rheumatology (Oxford) 2022; 61 (2): 490–501. doi: 10.1093/rheumatology/keab634.
79. Sun C, Xu G, Lin J. Comparison of IgG4-Related lymphadenopathy and multicentric Castleman’s disease: a retrospective study. Clin Lab 2018; 64 (10): 1671–1678. doi: 10.7754/Clin.Lab.2018.180421.
80. Izumi Y, Takeshita H, Moriwaki Y et al. Multicentric Castleman disease mimicking IgG4-related disease: a case report. Mod Rheumatol 2017; 27 (1): 174–177. doi: 10.3109/14397595.2014.985356.
81. Yoshimi R, Nakajima H. Idiopathic multicentric Castleman’s disease as a Mimicker of IgG4-related disease. Intern Med 2025; 64 (12): 1788–1790. doi: 10.2169/internalmedicine.4793-24.
82. Qin Y, Shang L, Wang Y et al. Immune profile differences between IgG4-related diseases and primary Sjögren’s syndrome. J Inflamm Res 2025; 18 : 911–923. doi: 10.2147/JIR.S471266.
83. Otani Y, Shimura T, Nogaki T et al. Differentiation between IgG4-related Mikulicz disease and Sjögren’s syndrome: a review case report and literature review. Medicine (Baltimore) 2022; 101 (52): e32617. doi: 10.1097/MD.0000000000032617.
84. Yu L, Shi M, Cai Q et al. A novel predictive model for idiopathic multicentric Castleman disease: the international Castleman disease consortium study. Oncologist 2020; 25 (11): 963–973. doi: 10.1634/theoncologist.2019-0986.
85. Covelli C, Carosi I, Graziano P et al. When idiopathic retroperitoneal fibrosis mimics Castleman disease: a challenging differential diagnosis. BMJ Case Rep 2022; 15 (4): e248051. doi: 10.1136/bcr-2021-248051.
86. Fichtl J, Třeška V, Vodička J et al. Castlemanova choroba – neobvyklý nález při operaci tumoru retroperitonea u mladého nemocného. Rozhl Chir 2016; 95 (2): 91–94.
87. Szturz P, Plank L, Křístek J et al. Castlemanova choroba v obrazech. Postgrad Med 2014; 16 (1): 81–88.
88. Kazakov DV, Fanburg-Smith JC, Szster S et al. Castleman disease of the subcutis and underlying skeletal mucle: report of 6 cases. Am J Surg Pathol 2004; 28 (5): 569–577. doi: 10.1097/00000478-200405000-00002.
89. Moláček J, Treska V, Skalický V. et al. Unicentric form of Castleman’s disease, pitfalls of diagnosis and surgical treatment. Front Oncol 2023; 13 : 1057683. doi: 10.3389/fonc.2023.1057683.
90. Škach J, Vytiska J, Gaalová R et al. Castlemanova choroba imitující tumor perikardu. Kazuistiky v alergologii, pneumologii a ORL 2014; 11 (1): 3–7.
91. Čéška F, Ferko A, Jon B et al. Pancreatic Castleman disease treated with laparoscopic distal pancreatectomy. Hepatobiliary Pancreat Dis Int 2013; 12 (3): 332–334. doi: 10.1016/s1499-3872 (13) 60053-3.
92. Zavázalová Š, Jirák P, Syrůček M et al. Castlemanova choroba – mimicking a malignant lymphoma. Otorinolaryngol Foniatr 2014; 63 (4): 246–250.
93. Kazakov D, Morisson C, Plaza JA et al. Sarcoma arising in hyaline-vascular Castleman disease of skin and subcutis. Am J Dermatopathol 2005; 27 (4): 327–332. doi: 10.1097/01.dad.0000171606.55810.86.
94. Cibičková L, Soukup T, Bradna P. et al. Asociace revmatoidní artritidy a Castlemanovy choroby. Čes Revmatol 2005; 3 (3): 106–109.
95. Camprt V. Monitor aneb nemělo by vám uniknout, že už se ví, jak diagnostikovat idiopatickou multicentrickou Castlemanovu chorobu. Čes-slov Patol 2023; 59 (3): 89–95.
96. Ciferská H, Vachek J. Uzlinové syndromy – zaměřeno na Castlemanovu chorobu. Medicína po promoci 2021; 22 (3): 222–231.
97. Kufa J, Novotná J Lukášovíá M. et al. Raritní granulomatózní procesy. Transfuze Hematol Dnes 2024; 30 (2): 122–130. doi: 10.48095/cctahd2024prolekare.cz11.
98. Jakubíková M. Autoimunitní onemocnění periferního nervového svstému. Generalizovaná forma myasthenia gravis a jiné autoimunity na společném imunopatogenetickém pozadí interleukinu-6. Autoimunity nervového systému v kazuistikách II. 1. vyd. Praha: EEZY Publishing, s.r.o 2023 : 143–149.
99. Adam Z, Řehák Z, Boichuk I et al. Léčba Castlemanovy choroby z pohledu roku 2026. Klin Onkol 2026; 39 (2): 86–98. doi: 10.48095/ccko202686.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2026 Číslo 3
- Brno opět přivítá onkology a nelékařské zdravotnické pracovníky
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Fixní kombinace tramadol/paracetamol je doporučenou volbou v léčbě chronické bolesti v ordinaci praktického lékaře
Najčítanejšie v tomto čísle
- Rozdíly v zobrazení Erdheimovy-Chesterovy choroby FDG-PET/CT, NaF-PET/CT vyšetřením a scintigrafií skeletu. Ústup nemoci po léčbě kladribinem s cyklofosfamidem a anakinrou
- Histiocytární choroby dospělých – pestrost jejich klinických projevů vyžaduje spolupráci lékařů mnoha oborů
- Druhá a vyšší linie systémové léčby karcinomu endometria – zkušenosti z reálné klinické praxe
- Chronicky zvýšená teplota či horečka s nevelkou lymfadenopatií může být Castlemanova choroba, pokud se neprokáže maligní, autoimunitní anebo infekční příčina lymfadenopatie