
Klasifikace zárodečných variant identifikovaných při genetickém vyšetření nádorové predispozice – konsenzus konzorcia CZECANCA
Classification of germline variants identified in cancer predisposition genetic testing – consensus of the CZECANCA consortium
Background: Hereditary cancer syndromes are an important subset of malignant cancers caused by pathogenic variants in one of many known cancer predisposition genes. Diagnosis of cancer predisposition is based on genetic testing using next-generation sequencing. This allows many genes to be analysed at once, increasing the number of variants identified. The correct classification of the variants found is essential for the clinical interpretation of genetic test results.
Purpose: The aim of this study is to summarise the rules for classifying identified variants within individual laboratories and to present the process for creating a common classification. In the Czech Republic, the sharing of identified genetic variants and the development of their consensus classification among national laboratory diagnostic communities is carried out within the Czech Cancer Panel for Clinical Application (CZECANCA) consortium of scientific and diagnostic oncogenetic laboratories. Consensus for variant classification follows a defined protocol. Sharing the results and consensus classification accelerates and refines the release of genetic test results, harmonises results between laboratories and thus contributes to improving the care of individuals at high risk of cancer and their relatives.
Keywords:
genetic testing – massively-parallel sequencing – hereditary neoplastic syndromes – clinical relevance – variant classification – national consensus
Autori:
M. Janatová 1; Š. Chvojka 2; E. Macháčková 3; J. Soukupová 1; P. Zemánková 1,4; P. Nehasil 1,4,5; T. Zavoral 6; L. Hrušková 7; K. M. Kozáková Janíková 8 9; F. Lhota 2; S. Tavandzis 10; P. Kleiblová 1,11; Z. Kleibl 1; Czecanca Konzorcium
Pôsobisko autorov:
Ústav lékařské bio chemie a laboratorní dia gnostiky, 1. LF UK a VFN v Praze
1; Centrum lékařské genetiky a reprodukční medicíny, Gennet, Praha
2; Oddělení epidemiologie a genetiky nádorů, MOÚ Brno
3; Ústav patologické fyziologie, 1. LF UK a VFN v Praze
4; Klinika pediatrie a dědičných poruch metabolizmu 1. LF UK a VFN v Praze
5; Ústav lékařské genetiky, LF v Plzni UK a FN Plzeň
6; GHC Genetics, s. r. o., Praha
7; Ústav lékařské genetiky, LF UP a FN Olomouc
8; Genetická laboratoř PRONATAL, PRONATAL s. r. o., Praha
9; Oddělení lékařské genetiky, Laboratoře AGEL a. s., Nový Jičín
10; Ústav bio logie a lékařské genetiky, 1. LF UK a VFN v Praze
11
Vyšlo v časopise:
Klin Onkol 2023; 36(6): 431-439
Kategória:
Přehled
doi:
https://doi.org/10.48095/ccko2023431
Súhrn
Východiska: Hereditární nádorové syndromy tvoří významnou podskupinu zhoubných nádorových onemocnění způsobených patogenními variantami v některém z mnoha známých nádorových predispozičních genů. Diagnostika nádorové predispozice je založena na genetickém testování pomocí sekvenování nové generace. To umožňuje analýzu mnoha genů najednou, nicméně zároveň se zvyšuje počet identifikovaných variant. Správná klasifikace nalezených variant je zásadní pro klinickou interpretaci výsledků genetického testování.
Cíl: Cílem práce je shrnutí pravidel pro klasifikaci identifikovaných variant v rámci jednotlivých pracovišť a představení procesu tvorby společné klasifikace. Sdílení nalezených genetických variant a tvorba jejich konsenzuální klasifikace v rámci národních laboratorně diagnostických komunit probíhá v ČR v rámci konzorcia Czech Cancer Panel for Clinical Application (CZECANCA) sdružujícího výzkumné a diagnostické onkogenetické laboratoře. Tvorba konsenzu pro klasifikaci variant probíhá podle definovaného protokolu. Sdílení výsledků a konsenzuální klasifikace zrychluje a zpřesňuje vydávání výsledků genetického testování, harmonizuje výsledky mezi laboratořemi a přispívá tak ke zkvalitnění péče o jedince ve vysokém riziku vzniku nádorových onemocnění a jejich příbuzné.
Klíčová slova:
genetické testování – masivně paralelní sekvenování – hereditární nádorové syndromy – klinická relevance – klasifikace variant – národní konsenzus
Úvod
Hereditární nádorové syndromy jsou heterogenní skupina dědičných onemocnění charakterizovaných vznikem různých typů tumorů u nosičů vzácných germinálních mutací v nádorových predispozičních genech. Nejčastěji se jedná o tumor supresorové geny, jejichž produkty se negativně podílejí na regulaci mitotických signálů a buněčného cyklu, aktivují apoptózu nebo jsou zapojeny do oprav poškození DNA [1]. Nádorová onemocnění vznikající na podkladě dědičné predispozice představují 5–10 % případů všech onkologických onemocnění, nicméně mezi diagnózami se zastoupení dědičných forem liší a např. u karcinomu ovaria představují až 25 % [2]. Pro nastavení správné péče o pacienty s hereditárními nádorovými onemocněními a jejich příbuzné je nezbytná správná identifikace příčinné patogenní varianty, která má jasný prognostický, ale často i prediktivní význam [3]. Za klinicky významné v současnosti považujeme varianty/geny, u nichž riziko vzniku onemocnění (odds ratio – OR) přesahuje dvojnásobek rizika běžné populace (OR > 2). Geny/varianty se střední penetrancí jsou spojeny s rizikem v intervalu OR = 2–4. Přesahuje-li riziko vzniku onkologického onemocnění OR ≥ 4, hovoříme o genech s vysokou penetrancí. Termín „penetrance“ prakticky charakterizuje podíl jedinců, kteří vyvinou určité nádorové onemocnění z celkového počtu všech nosičů hodnocené varianty (box 1).
Box 1.
Pro správnou interpretaci celkového (celoživotního) rizika je nezbytné konkretizovat OR s ohledem na výskyt sledovaného nádorového onemocnění v běžné populaci. Riziko vzniku karcinomu prsu u nosiček patogenních variant v genu BRCA1 je OR = 6–8. Protože celoživotní riziko vzniku karcinomu prsu běžné populace žen ve věku 50 let dosahuje v ČR přibližně 10 %, vyplývá z toho, že celoživotní riziko stejně staré nosičky patogenní varianty v genu BRCA1 je 60–80 %. Muž, nosič patogenní varianty v genu BRCA1 má ve srovnání s nenosičem riziko pro vznik karcinomu prsu mnohem vyšší OR = 20–30. Protože však celoživotní riziko vzniku karcinomu prsu v běžné populaci mužů je 0,1 %, celoživotní riziko vzniku karcinomu prsu u mužských nosičů BRCA1 mutace bude stále „pouze“ 2–3 %.
V současné době všechny klinicko-genetické laboratoře u nás využívají ke genetickému testování pacientů s hereditárními nádorovými onemocněními sekvenování nové generace (next generation sequencing – NGS). NGS umožňuje identifikaci variant v mnoha cílových úsecích DNA v definovaném souboru vybraných genů (při panelovém sekvenování), nebo v celém exomu (souboru všech genů kódujících proteiny při celoexomovém sekvenování), či v genomu (při celogenomovém sekvenování analyzujícím veškerou jadernou DNA). Technologie NGS je založena na masivně paralelním sekvenování, tedy analýze mnoha (milionů) cílových úseků najednou [4]. Neustálé zvyšování kapacity a zlevňování NGS umožňuje jednoduše rozšiřovat spektrum vyšetřovaných genů. To se ukázalo jako významný krok k zjištění mnohem rozmanitějšího fenotypového spektra nosičů patogenních variant v nádorových predispozičních genech, než tomu bylo v dobách testování jednotlivých genů vycházejícího z analýz omezeného počtu typických rodin nosičů patogenních variant v hlavních predispozičních genech [5]. Rozšíření panelů tak dále přispívá ke zvyšování počtu genů s potvrzenou klinickou relevancí [3].
Na druhou stranu s tím souvisí také neustálé zvyšování počtu identifikovaných genetických variant, které zahrnují všechny odchylky od referenční sekvence. Nalezené varianty mohou být jasně klinicky hodnotitelné, ale často se jedná o varianty s nejasným klinickým významem (variant of uncertain significance – VUS), u kterých nejsme schopni kvalifikovaně potvrdit, že se tyto varianty podílejí na vzniku onemocnění, ani vyloučit, že se na vzniku onemocnění nepodílejí. Přítomnost VUS značně ztěžuje klinickou interpretaci výsledků NGS a výtěžnost genetického vyšetření [6].
Robustní, rychlá a přesná analýza variant, jejich správná klasifikace a interpretace asociovaného rizika onemocnění má zásadní význam pro efektivní poskytování výsledků genetického testování. Konsenzuální klasifikace pak zajišťuje konzistenci výstupů genetického testování mezi pracovišti v celé republice. Snaha o konzorciální spolupráci na národní úrovni probíhá v mnoha zemích [7–10]. Koncept sdílení variant je začleněn do tzv. best-practice guidelines [11]. Vytvoření procesu jednotné klasifikace variant na úrovni spolupráce pracovišť sdružených v konzorciu Czech Cancer Panel for Clinical Application (CZECANCA) v rámci národní laboratorně diagnostické komunity v ČR je tedy důležitým příspěvkem ke standardizaci výsledků genetického testování nádorové predispozice.

Konzorcium CZECANCA
Konzorcium CZECANCA je dobrovolné uskupení pracovišť zabývajících se klinickou onkogenetikou. K diagnostice nádorové predispozice využívají spolupracující pracoviště převážně panel CZECANCA, který vznikl v roce 2015 jako diagnostický nástroj spolehlivého a rentabilního NGS vyšetření širokého spektra genů u pacientů s nádorovou predispozicí v ČR [12,13]. Panel CZECANCA prošel v průběhu doby vývojem ovlivněným rozšiřujícím se portfoliem predispozičních genů i změnami v technologické přípravě. V současné podobě obsahuje 226 nádorových predispozičních a kandidátních genů a lze jej rozšířit i o analýzu polygenního rizika, jakmile vyvstane klinická poptávka [14]. Použití širokého panelu umožňuje charakterizovat nové nádorové predispoziční geny či možnou manifestaci již známých nádorových predispozičních genů u pacientů s doposud neasociovanými typy onkologických onemocnění. Použití jednotného panelu není podmínkou členství v konzorciu CZECANCA, na rozdíl od sdílení anonymizovaných NGS dat analyzovaných osob doplněných o základní klinicko-patologické charakteristiky sloužící pro tvorbu společné konzorciální databáze.
Postup NGS analýzy je mezi pracovišti v rámci konzorcia do značné míry sjednocen, což umožňuje i sjednocovat a standardizovat výsledky genetických analýz. Bioinformatická analýza NGS dat pro identifikaci variant je pro diagnostické účely realizována jednotlivými pracovišti dle vlastních postupů, avšak pro tvorbu společné frekvenční databáze CZECANCA je prováděna dle jednotného publikovaného postupu [12,13]. Databáze neobsahuje žádná individuální data jednotlivých pacientů, ale pouze údaje o kumulativní frekvenci nosičů dané varianty v souborech pacientů s různými onkologickými diagnózami v zapojených diagnostických centrech, v souborech kontrol z mezinárodních databází a v databázích kontrolních osob z české populace.
Databáze variant je pravidelně aktualizována v Laboratoři onkogenetiky Ústavu lékařské biochemie a laboratorní diagnostiky (ÚLBLD) 1. LF UK a VFN v Praze, kde rovněž probíhá analýza populačně specifických kontrol, která je důležitým prvkem pro detekci národních (populačně specifických) genetických variant. Databáze je k dispozici všem spolupracujícím centrům pro jejich interní využití při hodnocení variant nalezených u jimi vyšetřovaných pacientů.
Kromě klinických pracovišť se na komplexním hodnocení variant podílí i laboratoře výzkumné v rámci spoluprací a grantových projektů. Výzkumná část Laboratoře onkogenetiky ÚLBLD 1. LF UK a VFN v Praze zajišťuje hodnocení variant ovlivňujících sestřih pomocí NGS analýzy mRNA. Pod vedením Oddělení biologie nádorové buňky Ústavu molekulární genetiky AV ČR probíhají studie zaměřené na charakterizaci vybraných VUS pomocí in vitro funkčních analýz [15–17]. Aktivity národního konzorcia jsou koordinovány s mezinárodními konzorcii (ENIGMA, CIMBA, BCAC, OCAC), což je důležitým předpokladem pro identifikaci rizik spojených s nosičstvím velmi vzácných variant i pro konsenzuální hodnocení variant ve světě.
Konzorcium CZECANCA organizuje pravidelná setkání svých členů od roku 2016. Od roku 2021 probíhají setkání společně s Pracovní skupinou onkogenetiky Společnosti lékařské genetiky (SLG) ČLS JEP, sdružující členy SLG se zájmem o tuto problematiku. Program setkání se v průběhu času proměňuje. Zatímco v počátcích byly řešeny hlavně otázky spojené s laboratorním zpracováním vzorků a bioinformatickou analýzou, v současné době dominují témata klasifikace variant, rozsahu reportovaných genů a souvisejících klinických doporučení. Zapojená pracoviště se tak společně podílejí na vytvoření národního konsenzu týkajícího se jednotné klasifikace identifikovaných variant a rozsahu genů reportovaných indikujícím lékařům při vyšetření nádorové predispozice a na přípravě klinických doporučení pro nosiče klinicky relevantních germinálních variant.
Klasifikace nalezených germinálních variant
Bioinformatická analýza primárních NGS dat z panelu CZECANCA odhalí přítomnost několika set až tisíce variant u každého pacienta, v nichž je nezbytné vyhledat nebo vyloučit přítomnost případné patogenní varianty, zodpovědné za vznik onemocnění. Proces filtrace na soubor kandidátních variant se obvykle označuje jako prioritizace a zohledňuje řadu aspektů, především vyloučení častých (tedy funkčně i klinicky bezvýznamných) variant vyskytujících se v dostupných mezinárodních databázích či v souborech českých populačních kontrol. Po prioritizaci se počet dále hodnocených variant zúží na jednotky až nízké desítky kandidátních variant.
Dalším krokem je klasifikace kandidátních variant v rámci laboratorní zprávy určené klinickému genetikovi, zajišťující, že ve výsledné zprávě pro ošetřujícího lékaře budou reportovány (byly-li nalezeny) pouze klinicky relevantní varianty modifikující riziko vzniku onemocnění, jeho léčbu či péči o pacienta, případně jeho příbuzné. Klasifikace nalezených variant je stěžejním, a i jedním z nejobtížnějších kroků procesu genetického vyšetření. Klasifikovány jsou nejen standardně anotované jednonukleotidové záměny a krátké delece/inzerce, ale i středně dlouhé inzerce/delece (> 50 bp) a tzv. copy number variations (CNV) – intragenové a intergenové delece/duplikace. Klasifikace nalezených variant probíhá samostatně v rámci jednotlivých pracovišť v ČR na základě doporučení American College of Medical Genetics (ACMG) poskytujících pro jednotnou klasifikaci variant obecný rámec a srovnání s databází ClinVar (viz dále).
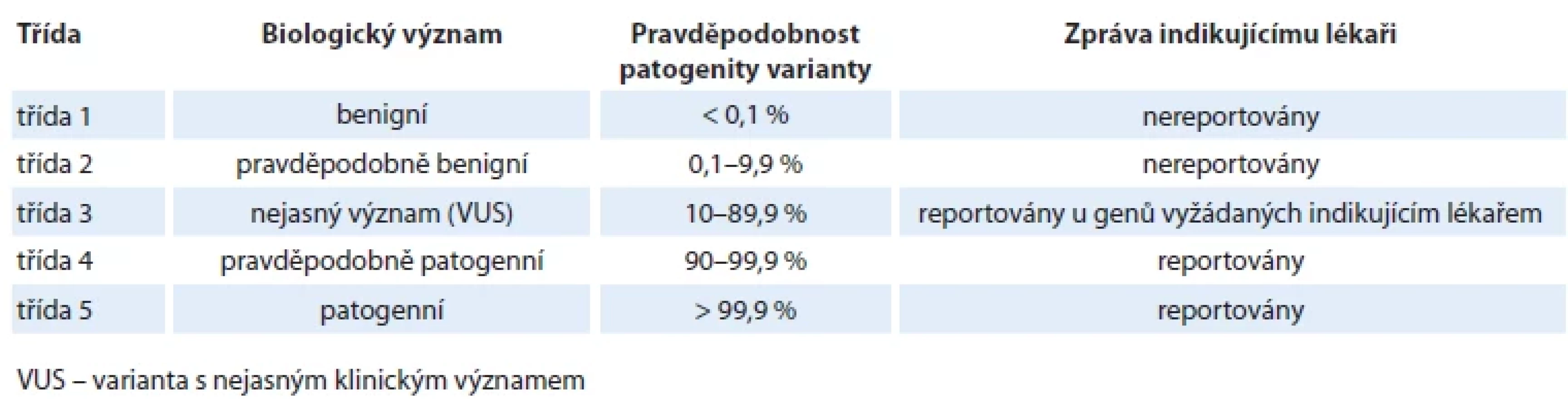
Základem současné klasifikace variant je tedy pětitřídní klasifikace dle ACMG (tab. 1), která rozděluje varianty na základě pravděpodobnosti patogenity – příčinné souvislosti přítomnosti varianty se vznikem konkrétního fenotypu (v našem případě s rozvojem konkrétního typu onkologického onemocnění) [18,19].
Varianty třídy 4 a 5 zahrnují varianty s pravděpodobností patogenity ≥ 90 % resp. ≥ 99,9 %. Varianty třídy 4 a 5 v klinicky relevantních nádorových predispozičních genech (geny s vysokou až střední penetrancí) jsou zodpovědné za riziko vzniku konkrétního onkologického onemocnění a jsou vždy reportovány indikujícímu lékaři. Z klinického úhlu pohledu není žádný rozdíl mezi nosičem varianty třídy 4 a třídy 5. Na základě mezinárodních a národních doporučení by měly být reportovány varianty třídy 4/5 i v některých dalších klinicky významných testovaných genech, které nesouvisí s primárním účelem testování (tzv. neočekávaný nález). Jedná se o varianty, jejichž přítomnost ovlivňuje péči o nosiče těchto variant či jejich rodiny, kteří z této znalosti mohou profitovat. Reportování neočekávaných nálezů je předmětem živých diskuzí a proto zatím neprobíhá zcela konsenzuálním způsobem (box 2).
Varianty třídy 3 představují VUS. Pravděpodobnost jejich patogenity se pohybuje v rozmezí 10–90 %. S ohledem na velmi široký interval patogenity, tedy i vysokou míru neurčitosti, nejsme schopni u VUS určit jejich klinický význam pro péči u pacientů a jejich příbuzných. Obvykle jsou VUS reportovány pouze v genech s jasným vztahem k fenotypu testovaného jedince.
(Pozn. odlišný přístup k významu VUS variant mohou mít analýzy somatických variant, protože jejich přítomnost ve vyšetřovaných vzorcích nádoru může reprezentovat přítomnost varianty pro cílenou léčbu).
Varianty třídy 1 a 2 (pravděpodobnost patogenity < 10 % pro třídu 2, < 0,1 % pro třídu 1) nemají žádný klinický význam a reportovány nejsou.
Varianty v genech, jejichž proteinové produkty se podílejí na metabolizmu léčiv (např. DPYD) [20], jsou klasifikovány jako „drug response“. Varianty, které poškozují funkci takového kódovaného proteinu a mají vliv na vznik závažné toxicity po podání příslušného léku, jsou reportovány indikujícímu lékaři.
Box 2.
U nosičů varianty třídy 4/5 v klinicky relevantních nádorových predispozičních genech je možné se vyjádřit pouze k riziku (rizikům) vzniku onkologických onemocnění, která jsou prokazatelně spojena s jejich s nosičstvím, a která jsou ovlivněna penetrancí dané varianty:
Přítomnost varianty třídy 5 v genu BRCA1 znamená pro nosičku > 99,9% pravděpodobnost, že její celoživotní riziko vzniku karcinomu prsu dosáhne 80 %.
Přítomnost varianty třídy 5 v genu CHEK2 znamená pro nosičku > 99,9% pravděpodobnost, že její celoživotní riziko vzniku karcinomu dosáhne 30 %.
Přítomnost varianty třídy 5 v genu DPYD vypovídá s > 99,9% pravděpodobností o riziku vzniku toxicity po podání fluoropyrimidinů, ale nevypovídá nic o riziku onkologického onemocnění, tedy není z hlediska karcinomu prsu klinicky významná, ale může ovlivnit péči o pacienta (v případě jeho léčby).
Přítomnost varianty třídy 5 v genu BLM znamená, že nosič dvou takovýchto variant (homozygot nebo složený heterozygot) vyvine s > 99,9% pravděpodobností Bloomův syndrom (vzácné autozomálně recesivní onemocnění), ale podíl nosičství jedné této varianty na vzniku karcinomu prsu je zcela nejasný a s onemocněním nemusí souviset vůbec.
ACMG doporučení zohledňují řadu parametrů (např. vliv varianty na strukturu a funkci kódovaného proteinu, frekvence varianty v populaci, in silico predikce, funkční analýzy, databáze, publikace), přičemž váha těchto parametrů pro konečnou klasifikaci varianty se liší [18]. Pro některé geny byla vytvořena modifikovaná ACMG doporučení, jež zohledňují jejich specifika (např. TP53, ATM) [21,22]. Jakkoliv jsou ACMG kritéria pro klasifikaci variant jednotící platformou se snahou o objektivizaci klasifikace genetických nálezů, subjektivní hodnocení klasifikačních parametrů může vést k diskrepantnímu hodnocení variant mezi jednotlivými pracovišti.
Nejpoužívanějším podkladem pro klasifikaci je veřejně dostupná databáze ClinVar shromažďující varianty identifikované různými klinickými a výzkumnými laboratořemi z celého světa doplněné o komentář k jejich klasifikaci. Při využití hodnocení variant z databáze ClinVar je však potřeba mít na zřeteli spolehlivost hodnocení vyjádřenou počtem hvězdiček (*). Počet hvězdiček vyjadřuje míru relevance, která vzniká v poměrně složitém procesu hodnocení variant expertními panely nebo odbornými společnostmi/konzorcii (tab. 2).
Existují další zdroje, např. iniciativa ClinGen, jako nadstavba databáze ClinVar, poskytující navíc informaci o relevanci testovaných genů k fenotypu pacienta. Řada dalších databází a platforem (BRCA Exchange, LOVD, IARC) shromažďuje informace o nalezených variantách vč. jejich klasifikace nejen u pacientů s dědičnými nádorovými onemocněními.
Klasifikace variant dle jejich vlivu na funkci kódovaného proteinu je pouze první úrovní při interpretaci výsledků genetického vyšetření. Pro výslednou interpretaci varianty je vždy nutné individuálně zohlednit nejen vliv dané varianty na biologickou funkci proteinu, ale také vztah varianty k fenotypu, zahrnující penetranci, riziko (OR) a konkordanci (souvislost výskytu patogenní varianty s přítomností typického onkologického onemocnění). Penetrance variant třídy 4/5 se mezi geny výrazně liší (viz výše). Ovšem může se lišit i v rámci jednoho genu. Příkladem je „missense“ varianta v genu BRCA1: c.5096G>A, p. (Arg1699Gln), která je v databázi ClinVar vedena jako patogenní, nicméně celoživotní riziko vzniku karcinomu prsu u jejích nosičů dosahuje ~20 % (spadá do kategorie variant se střední penetrancí) [23] a je tak významně nižší než riziko spojené s ostatními jasně patogenními (a vysoce penetrantními) variantami v genu BRCA1 s celoživotním rizikem vzniku karcinomu prsu 60–80 % [24]. Rovněž je potřeba brát v potaz typ dědičnosti. Zatímco bialelické patogenní varianty v genu MUTYH jsou spojeny s vysokým rizikem (70–90 %) vzniku kolorektálního karcinomu (colorectal cancer – CRC), heterozygotní mutace v tomto genu zvyšují riziko CRC pouze mírně (6–13 %) [25]. Management nosičů se následně odvíjí od výše rizik spojených s nosičstvím identifikované varianty v kontextu osobní a rodinné anamnézy.
Tvorba národního konsenzu klasifikace germinálních variant
Klasifikace variant do tříd má zásadní dopad na péči o pacienty. Pro dosažení společného národního konsenzu v rámci konzorcia CZECANCA byla stanovena pevná pravidla. Varianty ve společné databázi jsou nejprve individuálně hodnoceny jednotlivými (osmi) pracovišti. Pro dosažení konsenzuální klasifikace je nutné, aby byly varianty hodnoceny alespoň třemi centry v rámci konzorcia. Konsenzus je stanoven přímo pokud: 1) varianta získá hodnocení s úplnou shodou, alespoň od 3 center; 2) pokud u varianty převažuje shodné hodnocení, alespoň od 4 center, a přitom s rozdílem klasifikace maximálně o 1 třídu ve smyslu 1/2 nebo 4/5. V případě diskrepantních hodnocení jsou varianty postoupeny k diskuzi komisi složené ze tří vybraných zástupců hodnotitelů variant z různých pracovišť, která se snaží dosáhnout společného konsenzu v hodnocení. Výsledný konsenzus u takových variant je podroben zpětné oponentuře všemi centry. V případě neshody v rámci komise a v případě diskrepantních hodnocení s rozdílem dvou a více tříd s hodnocením přecházející přes třídu 3, tedy rozdíl ovlivňující klinickou péči (např. třídy 2–4, 3–5) jsou varianty diskutovány plénem v rámci setkání konzorcia (schéma 1).





Výsledky národní klasifikace jsou pravidelně (~ 1× ročně) porovnávány s databází ClinVar a rozdílná hodnocení jsou posouzena komisí. V případě změny klasifikace na základě nejnovějších poznatků jsou informována všechna pracoviště a nové hodnocení upraveno v nejnovější verzi frekvenční databáze. V případě změny klasifikace varianty vedoucí ke změně péče o nosiče (reklasifikace variant třídy 3 na varianty třídy 4/5 nebo obráceně) je dodatečně vydána zpráva lékaři, který indikoval nosiče takovéto varianty ke genetickému testování. V současné době jsou konsenzuálně klasifikované varianty v rámci konzorcia CZECANCA vkládány do databáze ClinVar pouze epizodicky, kompletní sdílení variant je připravováno.
CZECANCA databáze verze 5.0 obsahuje výsledky vyšetření 10 480 pacientů s nádorovými onemocněními a 791 osob ze skupiny nenádorových kontrol. Celkem bylo nalezeno 77 812 různých sekvenčních variant zárodečné DNA. Z nich se 26 874 vyskytovalo v kódujících exonových a přilehlých intronových oblastech. Pro tvorbu konsenzu bylo doposud (k 31. 5. 2023) klasifikováno 20 genů asociovaných se syndromem hereditárního karcinomu prsu a ovaria a genů Lynchova syndromu: ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, EPCAM, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, STK11 a TP53. Hodnocení se účastnilo 8 pracovišť.
Ve výše uvedených genech bylo celkem přítomno 9 797 variant. Z nich se 3 428 vyskytovalo v kódujících exonových a přilehlých intronových oblastech. Alespoň jedním pracovištěm bylo hodnoceno 3 190 (93,1 %) variant. Alespoň třikrát bylo hodnoceno 2 017 (58,8 %) variant a 1 411 variant bylo hodnoceno pouze nedostatečným počtem pracovišť (graf 1).
Konsenzu bylo dosaženo přímo u 1 858 z 2 017 variant (92,1 %). Ke komisionálnímu hodnocení konsenzu bylo postoupeno 159 variant (7,9 %). Většinou se jednalo o nesouhlasné hodnocení ve smyslu tříd 1–2, 2–3, 1–3, nebo 4–5, které řešilo jednání hodnotící komise. V osmi případech se hodnocení lišilo o více než dvě třídy a přesahovalo třídu 3. Celkem jedenáct variant (osm přes třídu 3, tři bez shody v rámci komise) bylo diskutováno na společném setkání uživatelů.
V konečném konsenzu bylo klasifikováno 2 005 z 2 017 variant hodnocených dostatečným počtem členů (spolupracujících pracovišť) národního konzorcia. Dvanáct „missense“ variant nebylo zatím klasifikováno, vzhledem k diskordantním výsledkům funkčních analýz [16,17] a klasifikaci v databázi ClinVar.
V konečném konsenzu se podařilo přiřadit hodnocení s jasným klinickým významem (bez VUS) u 1 170 (58,4 %) variant. Z toho 771 (38,5 %) zařadit do tříd 1/2 a 399 (19,9 %) do tříd 4/5. Po zvážení všech dostupných informací ovšem stále zůstává 835 (41,6 %) variant klasifikovaných do třídy 3 (VUS) (graf 2).
Nejčastěji se hodnocení s nejasným klinickým významem vyskytují u vzácných „missense“ variant a dále u variant v přilehlých intronových oblastech mimo konsenzní sestřihová místa ±1/2, u nichž vliv na sestřih mRNA bez její analýzy lze na základě nepřesných predikčních algoritmů hodnotit pouze orientačně. Mezi geny s nejmenším zastoupením VUS patří geny BRCA1 (21,2 %) a BRCA2 (26,9 %), které jsou nejdéle spojovány s hereditární nádorovou predispozicí a jimž hodnocení je dlouhodobě věnováno velké úsilí. Naopak největší zastoupení VUS se vyskytuje v genech velkých (ATM – 58,1 %) a v genech, kterým je v rámci nádorové predispozice pozornost věnována kratší dobu (RAD51D – 63,2 %, CHEK2 – 58,2 %, NBN – 55,2 % a BARD1 – 54,2 %) (graf 3). Takové varianty představují problém v klinické interpretaci nálezů. V rámci spolupráce s mezinárodními konzorcii panuje obrovská snaha VUS varianty funkčně charakterizovat a následně klasifikovat do jasně klinicky interpretovatelných tříd 1/2 (bez klinického významu) nebo 4/5 (s jasným klinickým dopadem) [16,26,27].
Z 2 005 variant klasifikovaných CZECANCA konzorciem není přítomno v databázi ClinVar 284 variant (k 31. 5. 2023), vč. 80 klinicky významných variant klasifikovaných do tříd 4/5. Dalších 353 variant má konfliktní hodnocení, vč. 18 klinicky významných variant klasifikovaných do tříd 4/5. Ze zbylých 1 370 variant s jasnou klasifikací v databázi ClinVar jsme ve velké většině (99 %) dospěli ke stejné klasifikaci, pouze u 17 variant se klasifikace liší, v 15 případech na úrovni změny mezi třídami 2 a 3, tedy bez vlivu na péči o pacienta (graf 4).
Ve dvou případech se jednalo o klinicky významnou změnu mezi třídami 3 a 4: varianta v genu MLH1: c.1897-2A>G je hodnocena jako třída 4 expertní skupinou International Society for Gastrointestinal Hereditary Tumours (InSiGHT) z roku 2019 pouze na základě postižení konsenzního sestřihového místa, ovšem bez provedené analýzy RNA. Databáze ale obsahuje dalších 5 záznamů hodnotících variantu jako třída 3. To podporuje i naše analýza RNA, která sice potvrdila alteraci sestřihu MLH1 mRNA, avšak vedoucí k retenci tří nukleotidů způsobujících vložení jedné aminokyseliny do oblasti proteinu neobsahující žádnou známou funkční ani interakční doménu. Naopak, varianta MLH1: c.2252_2253dup je hodnocena skupinou InSiGHT z roku 2019 jako třída 3 bez dodatečných komentářů. Nicméně, databáze dále obsahuje další čtyři hodnocení jako třída 4 vč. odkazů na funkční analýzy potvrzující dopad na strukturu a funkci proteinu. Tyto příklady naznačují nezbytnost pečlivé kontroly hodnocení v databázi ClinVar.
Při klasifikaci variant je z hlediska frekvence variant v populaci nejčastěji využívaným zdrojem databáze GnomAD (The Genome Aggregation Database) databáze zahrnující exomová a genomová data z mezinárodních rozsáhlých sekvenačních projektů. V této frekvenční databázi není přítomno 688 dosud námi klasifikovaných variant, vč. 254 klinicky významných variant klasifikovaných jako třídy 4/5. Naopak frekvence některých populačně-specifických patogenních variant může být v GnomAD poměrně vysoká. Např. jasně patogenní varianta v genu CHEK2: c.1100del má v této databázi frekvenci > 0,2 % (databáze je obohacena o populace, kde je tato varianta nejčastější). Frekvence řady variant se tedy mezi populacemi může významně lišit, příkladem může být patogenní (founder) varianta v genu NBN: c.657_661del (označovaná také jako c.657del5), jejíž maximální frekvence v databázi GnomAD je v evropské populaci (mimo finské) 0,04 %, zatímco frekvence této varianty v české kontrolní populaci je 0,2 %.
Společný konsenzus je tak důležitým nástrojem i pro klasifikaci populačně specifických variant, které nelze najít v mezinárodních multipopulačních databázích nebo jsou v jednotlivých populacích zastoupeny s různou frekvencí.
Klasifikace variant v rámci konzorcia CZECANCA neustále probíhá, s každou další verzí frekvenční databáze přibývají hodnocení nových genů a variant. Změna klasifikací a následně i klinického významu bude jistě procházet dynamickým vývojem i nadále dle neustálého posouvání nejnovějšího poznání. Proto je nezbytná i průběžná zpětná kontrola již hotových hodnocení.
Závěr
Národní spolupráce v rámci konzorcia CZECANCA umožňuje komplexní přístup k vyšetření nádorové predispozice u nejčastějších solidních nádorů dospělého věku. Společný postup a sdílení výsledků při hodnocení germinálních variant zvyšuje uniformitu výsledků genetických analýz a snižuje výskyt chyb napříč diagnostickými centry v ČR a současně šetří čas hodnotících pracovníků. Konsenzuální přístup ke klasifikaci nalezených variant zkvalitňuje reportování laboratorních nálezů, které je nezbytné pro efektivní a optimální péči o pacienty s nádorovými onemocněními a jejich příbuzné.
Dalším cílem konzorciální spolupráce bude vytvořit národní konsenzus týkající se rozsahu genů reportovaných při vyšetření nádorové predispozice laboratořemi indikujícím klinickým genetikům. V rámci konzorcia shoda zatím nepanuje, nicméně je nezbytná pro další zlepšení a sjednocení genetického testování v rámci celé ČR. V procesu je také tvorba konsenzu týkajícího se klinických doporučení v péči o nosiče patogenních variant v nádorových predispozičních genech, jehož tvorba probíhá v rámci široké platformy klinických lékařů z různých oborů, kteří se podílejí na péči o tyto vysoce rizikové osoby. Jakkoliv došlo v oblasti genetického testování nádorové predispozice v ČR k obrovskému pokroku a v mezinárodním měřítku je naprosto srovnatelné, obrovskou výzvou bude naprosto nedostatečná kapacita ambulancí poskytujících péči nosičům patogenních variant v nádorových predispozičních genech.
Poděkování
Rádi bychom poděkovali všem členů konzorcia CZECANCA.
Podporující agentury a sponzoři:
Práce byla realizována za podpory grantů Agentury pro zdravotnický výzkum MZČR NU20-03-00285, NU20-03-00283, NU20-03-00016, NU20-09-00355, projektu MŠMT LM2023067 a projektů Karlovy Univerzity SVV260631 a COOPERATIO.
Zdroje
1. Kulkarni A, Carley H. Advances in the recognition and management of hereditary cancer. Br Med Bull 2016; 120 (1): 123–138. doi: 10.1093/bmb/ldw046.
2. Lhotova K, Stolarova L, Zemankova P et al. Multigene panel germline testing of 1333 Czech patients with ovarian cancer. Cancers (Basel) 2020; 12 (4): 956. doi: 10.3390/cancers12040956.
3. Turnbull C, Sud A, Houlston RS. Cancer genetics, precision prevention and a call to action. Nat Genet 2018; 50 (9): 1212–1218. doi: 10.1038/s41588-018-0202-0.
4. Bewicke-Copley F, Kumar EA, Palladino G et al. Applications and analysis of targeted genomic sequencing in cancer studies. Comput Struct Biotechnol J 2019; 17 : 1348–1359. doi: 10.1016/j.csbj.2019.10.004.
5. Walsh T, Lee MK, Casadei S et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010; 107 (28): 12629–12633. doi: 10.1073/pnas.1007983107.
6. Plon SE, Eccles DM, Easton D et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29 (11): 1282–1291. doi: 10.1002/humu.20880.
7. Fokkema IFAC, van der Velde K, Slofstra MK et al. Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data. Hum Mutat 2019; 40 (12): 2230–2238. doi: 10.1002/humu.23896.
8. Garrett A, Callaway A, Durkie M et al. Cancer Variant Interpretation Group UK (CanVIG-UK): an exemplar national subspecialty multidisciplinary network. J Med Genet 2020; 57 (12): 829–834. doi: 10.1136/jmedgenet-2019-106 759.
9. Wappenschmidt B, Hauke J, Faust U et al. Criteria of the German consortium for hereditary breast and ovarian cancer for the classification of germline sequence variants in risk genes for hereditary breast and ovarian cancer. Geburtshilfe Frauenheilkd 2020; 80 (4): 410–429. doi: 10.1055/a-1110-0909.
10. Tudini E, Andrews J, Lawrence DM et al. Shariant platform: enabling evidence sharing across Australian clinical genetic-testing laboratories to support variant interpretation. Am J Hum Genet 2022; 109 (11): 1960–1973. doi: 10.1016/j.ajhg.2022.10.006.
11. Acmg Board Of Directors. Laboratory and clinical genomic data sharing is crucial to improving genetic health care: a position statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 19 (7): 721–722. doi: 10.1038/gim.2016.196.
12. Soukupová J, Zemánková P, Kleiblová P et al. CZECANCA: CZEch CAncer paNel for Clinical Application – design and optimization of the targeted sequencing panel for the identification of cancer susceptibility in high-risk individuals from the Czech Republic. Klin Onkol 2016; 29 (Suppl 1): S46–54. doi: 10.14735/amko2016s46.
13. Soukupova J, Zemankova P, Lhotova K et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS One 2018; 13 (4): e0195761. doi: 10.1371/journal.pone.0195761.
14. Hovhannisyan M, Kleiblova P, Nehasil P et al. Polygenic risk score (PRS) and its potential for breast cancer risk stratification. Klin Onkol 2023; 36 (3): 198–205. doi: 10.48095/ccko2023198.
15. Stolarova L, Kleiblova P, Janatova M et al. CHEK2 germline variants in cancer predisposition: stalemate rather than checkmate. Cells 2020; 9 (12): 2675. doi: 10.3390/cells9122675.
16. Stolarova L, Kleiblova P, Zemankova P et al. ENIGMA CHEK2gether project: a comprehensive study identifies functionally impaired CHEK2 germline missense variants associated with increased breast cancer risk. Clin Cancer Res 2023; 29 (16): 3037–3050. doi: 10.1158/1078-0432.CCR-23-0212.
17. Kleiblova P, Stolarova L, Krizova K et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int J Cancer 2019; 145 (7): 1782–1797. doi: 10.1002/ijc.32385.
18. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17 (5): 405–424. doi: 10.1038/gim.2015.30.
19. Spurdle AB, Greville-Heygate S, Antoniou AC et al. Towards controlled terminology for reporting germline cancer susceptibility variants: an ENIGMA report. J Med Genet 2019; 56 (6): 347–357. doi: 10.1136/jmedgenet - 2018-105872.
20. Wörmann B, Bokemayer C, Burmeister T et al. Dihydropyrimidine dehydrogenase testing prior to treatment with 5-fluorouracil, capecitabine, and tegafur: a consensus paper. Oncol Res Treat 2020; 43 (11): 628–636. doi: 10.1159/000510258.
21. Fortuno C, Lee K, Olivier M et al. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat 2021; 42 (3): 223–236. doi: 10.1002/humu.24152.
22. Feliubadalo L, Moles-fernandez A, Santamarina-Pena M et al. A collaborative effort to define classification criteria for ATM variants in hereditary cancer patients. Clin Chem 2021; 67 (3): 518–533. doi: 10.1093/clinchem/hvaa250.
23. Moghadasi S, Meeks HD, Vreeswijk MP et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J Med Genet 2018; 55 (1): 15–20. doi: 10.1136/jmedgenet-2017-104560.
24. King MC. “The race” to clone BRCA1. Science 2014; 343 (6178): 1462–1465. doi: 10.1126/science.1251900.
25. Curia MC, Catalano T, Aceto GM. MUTYH: not just polyposis. World J Clin Oncol 2020; 11 (7): 428–449. doi: 10.5306/wjco.v11.i7.428.
26. Federici G, Soddu S. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J Exp Clin Cancer Res 2020; 39 (1): 46. doi: 10.1186/s13046-020-01554-6.
27. Monteiro AN, Bouwman P, Kousholt AN et al. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: best practices in functional analysis for clinical annotation. J Med Genet 2020; 57 (8): 509–518. doi: 10.1136/jmedgenet-2019-106368.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2023 Číslo 6
- Brno opět přivítá onkology a nelékařské zdravotnické pracovníky
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Fixní kombinace tramadol/paracetamol je doporučenou volbou v léčbě chronické bolesti v ordinaci praktického lékaře
Najčítanejšie v tomto čísle
- Klasifikace zárodečných variant identifikovaných při genetickém vyšetření nádorové predispozice – konsenzus konzorcia CZECANCA
- The endoplasmic reticulum and its signaling pathways – a novel target for multiple myeloma treatment
- Nová doporučení ESMO pro klinickou praxi u metastatického kolorektálního karcinomu – komentář ke změnám v systémové léčbě
- Paliativní radioterapie pokročilého karcinomu kůže ušního boltce