
Dabrafenib: nový inhibitor hyperaktivní kinázy B-RAF
Dabrafenib: the New Inhibitor of Hyperactive B-RAF Kinase
The B-RAF kinase is among major targets of biological therapy of cancer. B-RAF acts in the MAP kinase pathway, being activated by any of the RAS G-proteins. Hyperactive B-RAF is typically detected in chemoresistant and radioresistant malignant metastatic melanoma. In this study, we focus on the reversible ATP-competitive inhibitor dabrafenib (GSK-2118436), which is now in phase III clinical trial for use in subjects with various cancers expressing hyperactive B-RAF. Dabrafenib is selective for B-RAFV600E and B-RAFV600K (less for B-RAFV600D) over wild-type B-RAF. Thus, similarly to vemurafenib (Zelboraf), suggested is mandatory pre-screening for activating B-RAF mutations in the cancer tissue of each subject. Dabrafenib inhibits neoplastic growth at concentrations ≥ 53.8 nM in plasma, which corresponds to ≥ 30 mg/kg qd p.o., or to ≥ 3 mg/kg qd i.v. Most of the cancers expressing hyperactive B-RAF respond to dabrafenib treatment, but the complete response is only rarely achieved. Toxic side effects include skin lesions, pyrexia, frequent fatigue, nausea and pain. Resistance to dabrafenib is frequently developed via de novo RAS mutations, leading to the disease relapse. The RAS G-protein is capable of signaling downstream not only through B-RAF, but also through closely related C-RAF, which circumvents the effects of the B-RAF inhibitor. Thus, dabrafenib should not be prescribed to subjects with neoplasias that are positive for activating RAS mutations. Since B-RAF mutations alone cause only the formation of benign naevi, since the tumors frequently and quickly acquire resistance to B-RAF inhibitors, and because the B-RAF-inhibitor-mediated treatment outcomes are severely affected by changes in the activity and expression of a number of signaling molecules (among them PI3K/mTOR, PTEN, AKT, MEK, PDGFRβ), it can be anticipated that dabrafenib treatment should be suggested only as a part of combined therapy targeting simultaneously the other pathways responsible for cancer onset and progression.
Key words:
RAF1 – thyroid cancer – protein kinase inhibitors – antitumor agents – genetic predisposition to disease – neoplastic process – genetic markers – pharmacological biomarkers
Submitted:
29. 5. 2012
Accepted:
22. 8. 2012
Autoři:
P. Heneberg
Působiště autorů:
3. LF UK v Praze
Vyšlo v časopise:
Klin Onkol 2012; 25(5): 333-339
Kategorie:
Přehledy
Souhrn
Kináza B-RAF představuje jeden z hlavních cílů tzv. biologické léčby zhoubného bujení. Podílí se na aktivitě MAP kinázové dráhy, přičemž je spouštěna aktivací některého ze členů rodiny malých G proteinů rodiny RAS. Typickým nádorem exprimujícím hyperaktivní B-RAF je chemorezistentní a radiorezistentní maligní metastázující melanom. Tento příspěvek se věnuje vratnému ATP-kompetitivnímu inhibitoru dabrafenib (GSK-2118436), který již prochází III. fází klinických testů pro orální užití u pacientů s různými druhy nádorů exprimujícími hyperaktivní B-RAF. Dabrafenib je selektivní pro B-RAFV600E a B-RAFV600K, méně pro B-RAFV600D. K inhibici nemutované B-RAF kinázy však téměř nedochází, proto je podobně jako u vemurafenibu (Zelboraf) nutný genetický pre-screening nádorové tkáně na přítomnost aktivačních mutací v kináze B-RAF. Dabrafenib potlačuje růst nádorů při ≥ 53,8 nM koncentraci v plazmě, což odpovídá ≥ 30 mg//kg/den orálně, popřípadě ≥ 3 mg/kg/den intravenózně. Na dabrafenib reaguje většina nádorů exprimujících hyperaktivní B-RAF, kompletní odpovědi však bývá dosaženo jen zřídka. Toxické účinky zahrnují kožní léze, pyrexii, častou únavu, nevolnost a bolesti. Častý je vznik rezistence a následného relapsu onemocnění způsobených de novo mutacemi G proteinu RAS, který je schopen signalizovat nejen přes B-RAF, ale i přes blízce příbuzný C-RAF, a tím účinek inhibitoru obejít. Pacientům, v jejichž nádorech je aktivační mutace proteinu RAS zjištěna, by dabrafenib neměl být předepisován. Protože aktivační mutace v kináze B-RAF sama o sobě způsobuje pouze vznik benigních névů, protože nádor je schopen rychle získat na inhibitory kinázy B-RAF rezistenci a protože úspěšnost léčby inhibitory kinázy B-RAF ovlivňují změny v aktivitě a expresi celé řady dalších signálních molekul (PI3K/mTOR, PTEN, AKT, MEK, PDGFRβ aj.), lze předjímat, že užívání dabrafenibu bude nejefektivnější v kombinované terapii cílené na souběžné potlačení dalších signálních drah vedoucích k nádorovému bujení.
Klíčová slova:
RAF1 – karcinom štítné žlázy – inhibitory proteinkinázy – antitumorózní látky – genetická predispozice k nemoci – nádorové procesy – genetické markery – biomarkery farmakologické
Východiska
Kináza B-RAF představuje jeden z hlavních cílů tzv. biologické léčby zhoubného bujení, zejména pak v terapii metastázujícího melanomu. Tato kináza se podílí na aktivitě RAS/RAF/MEK/ERK dráhy, zkráceně nazývané též MAP kinázová dráha. Je spouštěna aktivací některého ze členů rodiny malých, na membránu vázaných G proteinů rodiny RAS. Aktivovaný protein RAS (v nádorech nejčastěji H-RAS, K-RAS či N-RAS) aktivuje rodinu proteinových serin/threoninových kináz RAF a MEK, což dále vyúsťuje v aktivaci MAP kináz (např. ERK či Elk), od kterých celá dráha odvozuje svůj široce rozšířený název. MAP kinázy se působením serin/threoninových kináz fosforylují, translokují se do buněčného jádra a stimulují genovou transkripci. Poměrně nedávno se podařilo prokázat, že celý systém funguje jako regulační smyčka, kdy v protisměru k MAP kinázové dráze působí jako zpětnovazebný mechanizmus dráha MAP kinázových fosfatáz [1].
Typickým nádorem, u kterého je deregulace MAP kinázové dráhy klíčovou pro jeho vznik, je maligní melanom. Jde o vysoce zhoubný novotvar odvozený z derivátů neurální lišty, jehož záchyt dlouhodobě roste ve většině industrializovaných zemí včetně České republiky [2]. Roku 2008 bylo v České republice zjištěno 1 029 případů metastatického melanomu u mužů a 1 005 případů u žen, přičemž jako příčina úmrtí byl maligní melanom uveden u 191 pacientů mužského a 158 pacientů ženského pohlaví [3]. Incidence a především mortalita melanomu je výrazně nižší než například u nádorů plic či tlustého střeva. Společenská významnost tohoto typu zhoubného bujení je však dána jeho nástupem již ve středním věku a s tím spojeným značným zkrácením doby dožití. Léčba melanomu byla donedávna omezena na chirurgickou resekci nádorového ložiska. Tento typ léčby je sice pro iniciální stadia melanomu velmi účinný a lze jej jednoznačně doporučit, nelze jej však použít v případě, kdy je melanom diagnostikován až v pokročilejším stadiu, kdy chirurgická léčba samozřejmě není schopna zabránit růstu již vzniklých metastatických ložisek. Konvenční přístupy pro léčbu pokročilých stadií maligního melanomu zahrnují adjuvantní imunoterapii (IFN-α a IL-2), popřípadě dakarbazin v kombinaci s multiferonem (dakarbazin je alkylující činidlo, multiferon je izolát různých typů lidského IFN-α). Podávání těchto léčiv je ale sporné s ohledem na to, že při poměrně silných vedlejších účincích dochází k odpovědi jen u méně než 20 % pa-cientů, délka této odpovědi se navíc počítá v řádu měsíců [4,5]. Alternativní přístup představuje CVD chemoterapie (cisplatina, vinblastin a dakarbazin kombinovaný s IFN-α a IL-2), BOLD terapie (bleomycin, vinkristin, lomustin a dakarbazin), popřípadě DBD/T kombinace (dakarbazin, karmustin, cisplatina a tamoxifen). Všechny tyto přístupy vedou k odpovědi u 18–64 % pacientů, avšak délka odpovědi na žádnou z těchto terapií nepřesahuje 12 měsíců [6]. Metastázující melanom je tedy stále považován za chemo - a radiorezistentní onemocnění a patří k nejobtížněji léčitelným diagnózám.
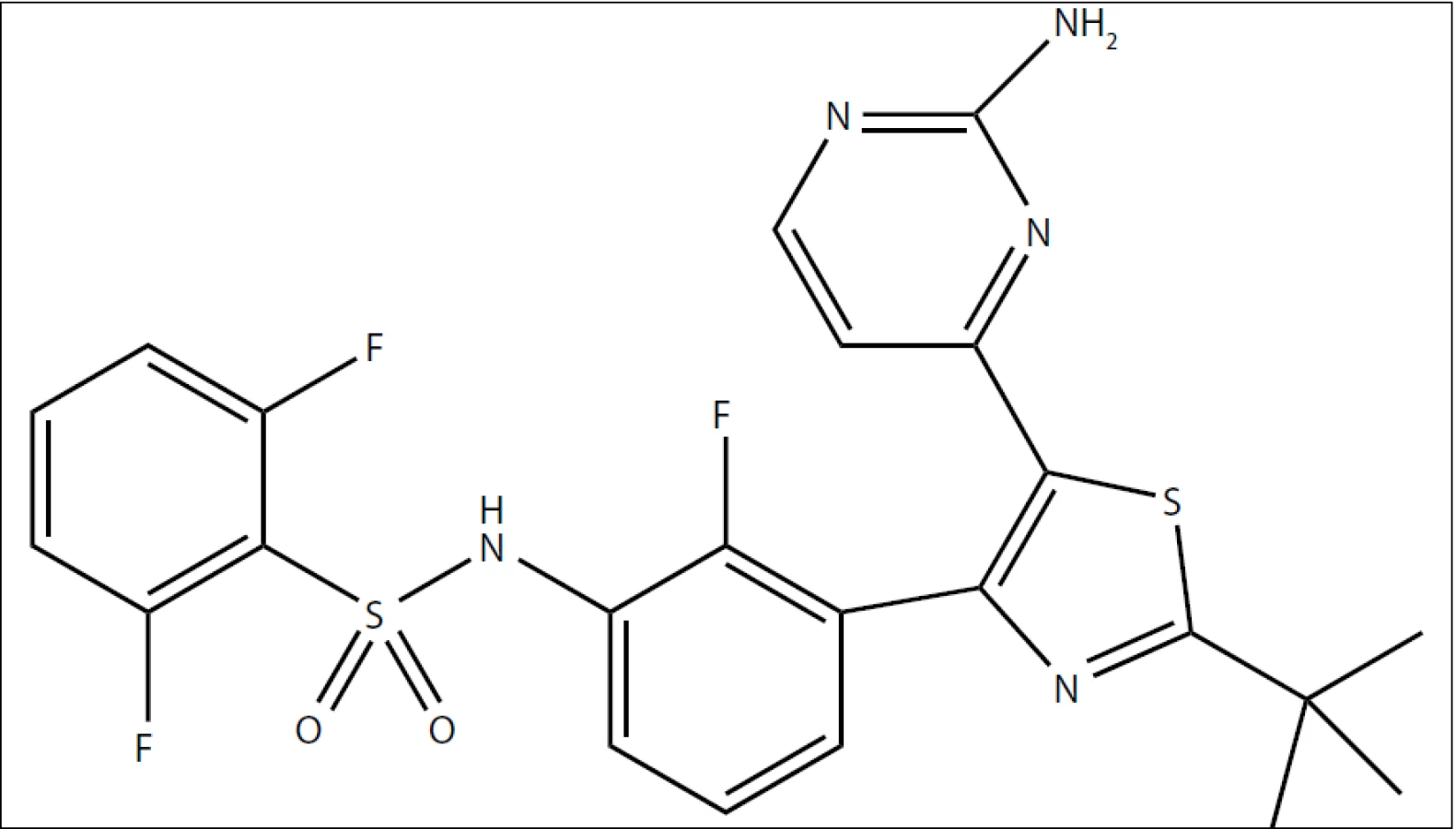
V poslední dekádě se objevilo hned několik pokusů o tzv. biologickou léčbu melanomu. Pod tento pojem se schovává několik na sobě nezávislých přístupů – imunoterapie zprostředkovaná monoklonální protilátkou proti CTLA-4 (ipilimumab), inhibice kinázy MEK (GSK1120212, trametinib DMSO), inhibice kinázy c-KIT (imatinib – účinný pouze u menšiny pacientů, jejichž melanom nese mutace v molekule c-KIT) a konečně inhibice kinázy B-RAF. Ačkoliv se na vzniku melanomu podílí poměrně velké množství genů [7], ve více než 80 % primárních melanomů se objevují mutace v dráze RAS/RAF/MEK//ERK (tab. 1), přičemž u maligních melanomů převažují právě aktivační mutace kinázy B-RAF (z 90 % B-RAFV600E). Přítomnost aktivačních mutací v kináze B-RAF je asociována s větší agresivitou melanomu a kratší dobou přežití od diagnózy [8,9]. Inhibitorů kinázy B-RAF se na trhu objevilo již několik, jejich první generace reprezentovaná sorafenibem (relativně širokospektrální inhibitor) byla stejně neúčinná jako konvenční chemoterapie či imunoterapie. Novější generace inhibitorů kinázy B-RAF reprezentovaná vemurafenibem (kterému jsme se pod označením PLX4032 v tomto časopise již věnovali podrobněji [2]) již sice překonává účinnost klasické chemoterapie či imunoterapie, avšak oproti dakarbazinu jde o zlepšení v řádech jednotek měsíců (celkové přežití o 5,3 měsíce; přežití bez progrese nemoci 1,6 měsíce [10]; nejnovější data prezentovaná na konferenci ASCO Meeting 2012 uvádějí delší přežití [11]). Nyní se do posledních fází klinických zkoušek dostal další z inhibitorů kinázy B-RAF zvaný dabrafenib (GSK-2118436), viz obr. 1. Dabrafenib vyvíjí společnost GlaxoSmithKline pro orální užití u pacientů s různými druhy nádorů s aktivačními mutacemi kinázy B-RAF, typicky právě pro pacienty s maligním melanomem.
![Přibližný podíl jednotlivých typů mutací molekul účastnících se MAP kinázové dráhy v melanomu [38].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/2a092432dceea61f132a21abf24f32aa.png)
Současné přístupy v České republice
Ještě než se budeme věnovat dabrafenibu podrobněji, připomeňme si, že Česká onkologická společnost ČLS JEP ve svých „Zásadách cytostatické léčby maligních onkologických onemocnění“ aktualizovaných v únoru 2012 [12] zmiňuje pouze konvenční chemoterapeutické přístupy kombinované s IFN-α a IL-2, popřípadě IFN-α samotný s tím, že v jednání o úhradě pojišťovnami je multiferon a ipilimumab (v průběhu recenzního řízení byla schválena částečná úhrada multiferonu v kombinaci s dakarbazinem pro léčbu pacientů klinického stadia IIB–III). Potenciální uplatnění biologické léčby není zmíněno. Dne 23. února 2012 byl v Evropské unii registrován k užívání i vemurafenib (pod názvem Zelboraf), který je v současnosti českým pacientům dostupný pouze v rámci individuálního dovozu, popřípadě v rámci klinické studie probíhající v pěti českých onkologických centrech [13]. Dabrafenib prozatím v EU registrován nebyl, ale vzhledem k tomu, že v zámoří je jeho testování již dokončováno, lze předjímat pokus o jeho registraci v blízké budoucnosti.
Dabrafenib – profil léčiva a jeho účinků
Dabrafenib je látka obsahující ve svém středu thiazolovou skupinu s navázaným benzensulfonamidem (na pozici 2) a 2-aminopyrimidinem (na pozici 5). Mechanizmus účinku spočívá v kompetici o vazebné místo na kináze B-RAF s ATP, inhibice je vratná. Jde o relativně selektivní látku (tab. 2) schopnou inhibovat aktivační mutanty kinázy B-RAF zahrnující jednak nejčastější B-RAFV600E, ale i méně časté B-RAFV600K a B-RAFV600D [14]. Inhibiční aktivita byla potvrzena i na buněčné úrovni na panelu 22 nádorových buněčných linií, kde ve všech případech dosahovalo IC50 dabrafenibu hodnot pod 100 nM [15]. V reakci na aplikaci dabrafenibu bylo zaznamenáno snížení fosforylace ERK, zastavení buněčné proliferace ve fázi G1 a následně buněčná smrt [16].
![Hodnoty IC<sub>50</sub> inhibitoru dabrafenib naměřené <em>in vitro</em> proti cílové mutantní B-RAF kináze a některým dalším příbuzným molekulám [38].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/b7642449ce954397f9f2de8fc91a1aef.png)
Při podání dabrafenibu pokusným myším s xenotransplantáty lidského melanomu, popřípadě nádoru tlustého střeva, bylo dosaženo snížení objemu nádoru při dávkách pohybujících se mezi 100 a 300 mg/kg (jedenkrát, popř. dvakrát denně); dávka řádově nižší (10 mg/kg) vedla k potlačení růstu xenotransplantátů. Pokud byl podán jen 1 mg/kg, došlo ke zpomalení růstu nádoru, při podání dávky 0,1 mg/kg nebyla zaznamenána žádná odpověď. Podobně jako u buněčných modelů popsaných výše i při podání dabrafenibu pokusným myším bylo zaznamenáno snížení fosforylace proteinů ERK a MEK [14–16]. Poměrně zásadním pozorováním je, že nebylo dosaženo dávky, která by vedla ke kompletní remisi nádoru, což samozřejmě autoři zpráv vzhledem ke svým konfliktům zájmů poněkud opomíjejí zdůraznit.
Na základě dávek použitých na myších modelech byly stanoveny i koncentrace použité pro první experimenty na lidských dobrovolnících. Bylo zjištěno, že potlačení fosforylace proteinu ERK (IC50) je dosaženo při koncentraci dabrafenibu v plazmě 35,5 nM (18,5 ng/ml), potlačení růstu nádoru (IC50) pak bylo dosaženo při koncentraci dabrafenibu v plazmě 53,8 nM (28 ng/ml), což odpovídá 30 mg/kg/den (orální podávání), popřípadě 3 mg/kg/den (intravenózně). Jak již vyplývá z rozdílu ve výše uvedených koncentracích dabrafenibu nutných k dosažení inhibice fosforylace proteinu ERK a k regresi nádoru, dosažení inhibice fosforylace proteinu ERK nelze použít jako marker predikující výsledek léčby. Experimentálně bylo zjištěno, že pouze v 70 % případů bylo snížení fosforylace proteinu ERK asociováno s regresí nádoru [17].
Ukončené a probíhající klinické studie
Klinická studie fáze I/II (NCT01072175) zkoumala kombinaci dabrafenibu (75 mg/den, orálně, ve dnech 1 a 15) s trametinibem DMSO (2 mg/den, orálně, ve dnech 2 až 25) u pacientů s aktivujícími záměnami v aminokyselině B-RAFV600. Bylo zjištěno, že kombinace s trametinibem nevede ke zvýšení incidence vedlejších účinků [18]. Studie NCT01231568 zkoumala vliv podávání vysokotukové diety a vliv velikosti částic dabrafenibu na jeho farmakokinetiku [19].
Ze zajímavějších výsledků dalších studií uveďme studii fáze I/II NCT00880321, která jako první prokázala pokles hladiny exprese markeru proliferace Ki67 přímo v nádorových ložiscích v odpovědi na podávání dabrafenibu. Zároveň v této studii byla jako metabolický marker použita [18F]fluorodeoxyglukóza-PET (FDG-PET), pomocí které je možno zjišťovat metabolickou odpověď organizmu na léčbu. U 11 ze 14 zkoumaných pacientů došlo k poklesu tohoto markeru (měřitelný pokles v širokém rozpětí mezi 5 a 100 %) [20]. Ke kompletní odpovědi po devíti týdnech došlo u tří pacientů z celkem 91 osob, kterým byl podáván dabrafenib v dávce 35–200 mg dvakrát denně. Dalších 35 pacientů vykázalo částečnou odpověď, u 38 osob bylo dosaženo stabilizace nemoci, u 15 nemocných nádor na léčbu významněji nereagoval a pokračoval v růstu [21]. V druhé části této klinické studie bylo zahrnuto více pacientů, kritériem jejich výběru byla záměna v aminokyselině B-RAFV600. Při léčbě dabrafenibem (150 mg dvakrát denně) bylo zjištěno, že dabrafenib je schopen potlačit velikost metastáz melanomu v mozkové tkáni o více než polovinu: jednalo se o 8 z 10 pacientů, z toho u tří pacientů došlo ke kompletnímu vymizení metastáz v mozku (detekováno pomocí magnetické rezonance za užití gadoliniové kontrastní látky). Identičtí pacienti vykazovali ústup metastáz i mimo dutinu mozkovou – u 8 z 10 pacientů došlo k alespoň 50% zlepšení, u tří pacientů došlo ke kompletnímu ústupu metastáz (v játrech, prsu a dutině břišní). Ve skupině pacientů s melanomem, se záměnou v B-RAFV600, ale bez metastáz v mozku došlo ke kompletní odpovědi u jedné osoby, k částečné odpovědi u deseti subjektů, stabilní nemoc byla zjištěna u osmi osob a progrese nemoci byla zjištěna u jediného pacienta [14,21]. Na pilotním souboru 23 pacientů bylo zjištěno, že k potlačení velikosti metastáz v mozkové tkáni následkem podávání dabrafenibu dochází pravděpodobně nezávisle na předchozích chirurgických zákrocích, radioterapii a stereotaktické radiochirurgii [22]. Předběžné výsledky analýzy 172 pacientů s melanomem IV. stadia s 0,5–4,0 cm metastázami v mozku zveřejněné na ASCO Meeting 2012 [23] potvrzují výše uvedený trend, kdy po podávání 150 mg dabrafenibu dvakrát denně bylo po osmi týdnech dosaženo odpovědi u 10 z 19 pacientů s melanomem nesoucím B-RAFV600E a u jednoho z pěti pacientů s melanomem exprimujícím B-RAFV600K (vše pacienti bez předchozí léčby mozkových metastáz). U pacientů, kteří před podáváním dabrafenibu podstoupili jiný způsob léčby mozkových metastáz, byla odpověď podobná – dosáhlo jí 8 z 15 B-RAFV600E a jeden ze dvou B-RAFV600K pacientů [23]. Souhrnně výše uvedené výsledky naznačují, že kompletní remise mozkových metastáz lze za použití monoterapie dabrafenibem dosáhnout jen ve výjimečných případech, typický je dočasný ústup metastáz následovaný opětovnou progresí nemoci [22].
Mezi nádory, na nichž byl účinek dabrafenibu testován, patří i papilární karcinom štítné žlázy pozitivní na záměnu v B-RAFV600. Z devíti pacientů nedosáhl kompletní remise žádný, dva pacienti měli částečnou odpověď na léčbu, šest dosáhlo stabilizace nemoci a u jednoho subjektu nemoc dále progredovala [14,21]. Je možné, že tato data naznačují nutnost ovlivnění některé z paralelních signálních drah specifických pro tento typ nádorů. U již několikrát zmíněného vemurafenibu je kupříkladu dosahováno dobré odpovědi na monoterapii vemurafenibem u pacientů s melanomy exprimujícími B-RAFV600E, avšak obdobná monoterapie u pacientů s nádory tlustého střeva exprimujícími B-RAFV600E nevede k významnějšímu zlepšení. Je-li nasazen vemurafenib v kombinaci s inhibitory EGFR, dosažená odpověď je několikanásobně lepší [24]. U pacientů s nádory tlustého střeva exprimujícími mutace v BRAFV600 neexistují data pro samotný dabrafenib, na ASCO Meeting 2012 byly však prezentovány první výsledky jejich terapie kombinací dabrafenibu a trametinibu. Úplné odpovědi nebylo dosaženo u žádného pacienta, částečné odpovědi bylo dosaženo u jednoho z 20 pacientů (5 %), stabilizace nemoci byla konstatována u 10 pacientů (50 %). Léčbu provázely silné vedlejší účinky, u 75 testovaných pacientů při dávce 150 mg dabrafenibu dvakrát denně a 2 mg trametinibu denně patřila mezi vedlejší účinky horečka (65 % pacientů), únava (47 %), nevolnost (44 %), zimnice (41 %), zvra-cení (31 %), zácpa (28 %), průjem (24 %) a vyrážka (21 %) [25].
Další významnější výsledky klinických studií dabrafenibu v současné době nejsou dostupné. V době psaní tohoto příspěvku probíhá hned několik studií fáze II (zaměřených na široké spektrum nádorů, jejichž společným jmenovatelem je přítomnost aktivačních mutací v kináze B-RAF) a jedna studie fáze III (NCT01227889), která se zaměřuje na srovnání účinků dabrafenibu s dakarbazinem u neléčených pacientů s melanomy III. nebo IV. stadia a s aktivačními mutacemi kinázy B-RAF. První výsledky zveřejněné na ASCO Meeting 2012 ukazují na zlepšené přežití bez progrese nemoci při podávání 150 mg dabrafenibu p.o. dvakrát denně oproti 1 000 mg/m2 IV q3w dakarbazinu. Zkoumaná kohorta byla poměrně rozsáhlá: 187 pacientů léčených dabrafenibem, 63 pacientů léčených dakarbazinem, přičemž přežití bez progrese nemoci bylo 5,1 vs 2,7 měsíce (dabrafenib vs dakarbazin) u pacientů s melanomem III. nebo IV. stadia, s odpovědí u 53 % pacientů léčených dabrafenibem a u 19 % pacientů léčených dakarbazinem [26].
Samotný dabrafenib je chráněn mezinárodním patentem [14,15], jeho ochrana byla prodloužena až do ledna 2030. Evropská (EP-02282636), japonská (JP-2011519940) a americká (US-20110172215) národní patentová přihláška na své přijetí zatím stále čekají.
Je nutno podotknout, že všechny výše zmíněné výsledky vycházejí výhradně z open-label studií, které byly ukončeny ve velmi nedávné době anebo ještě probíhají, a jejich výsledky byly proto zveřejněny pouze formou konferenčních příspěvků (na ASCO Meeting, AACR-NCI-EORTC Int. Congress atp.).
Toxicita a vedlejší účinky
V současné době nejsou k dispozici výsledky toxikologických studií na zvířecích modelech. Dabrafenib aktivuje CYP3A4-zprostředkované metabolické dráhy, jeho degradace vede k tvorbě několika vysoce aktivních látek (označovaných jako GSK-2285403, GSK-2298683 a GSK-2167542), z nichž některé přetrvávají v organizmu déle než dabrafenib samotný [21,27].
Lidští dobrovolníci obecně reportovali podobné problémy, jaké byly zaznamenány u inhibitoru s podobným mechanizmem účinku – u vemurafenibu [2]. Ze 184 pacientů zkoumaných v rámci studie NCT00880321 jeden pacient po podání léčiva upadl do bezvědomí a u dalšího pacienta ze zkoumaného souboru došlo k hyponatremii čtvrtého stupně. Velmi častými méně významnými vedlejšími efekty byly únava (40 % dobrovolníků), horečka (34 %), bolesti hlavy (30 %), nevolnost (30 %), kožní papilomy (24 %), hyperkeratéza (24 %), bolest v končetinách (23 %) a průjem (22 %) [21]. Kožní léze indukované dabrafenibem zahrnovaly hyperkeratotickou aktinickou keratózu (35 %), verruca vulgaris (32 %), Groverovu chorobu (24 %), seboroickou keratózu (27 %) a spinocelulární karcinom (14 %). Dále byl pozorován zvýšený výskyt akné, fotosenzitivita a změny ve struktuře a barvě vlasů [28]. Obdobné spektrum vedlejších účinků bylo recentně publikováno na základě předběžných výsledků studie NCT01227889 [26] a NCT01266967 [23]; poslední ze jmenovaných studií zaznamenala i dvě fatality. Podobně jako při podávání jiných inhibitorů kinázy B-RAF i po podání dabrafenibu byla v období 2–14 týdnů od zahájení podávání zaznamenána de novo tvorba spinocelulárních karcinomů, které byly dobře léčitelné klasickým chirurgickým přístupem. Závažnými vedlejšími účinky trpělo celkem 36 % osob, kterým byl dabrafenib podán – nejčastěji šlo právě o spinocelulární karcinomy (11 %), pyrexii (5 %) a infekce močového traktu (3 %). Žádný z dosud zaznamenaných vedlejších účinků neměl fatální následky [21], s výjimkou dvou případů v rámci studie NCT01266967 [23].
Zajímavý příspěvek na téma vedlejších účinků se objevil na ASCO Meeting 2012, kde J. S. Weber et al prezentovali data studie využívající kombinované terapie dabrafenibem a trametinibem. Tato kombinovaná terapie vykazovala snížené množství vedlejších účinků jak oproti monoterapii dabrafenibem (méně hyperproliferativních kožních lézí), tak oproti monoterapii trametinibem (méně případů závažných kožních vyrážek) [29].
Kontraindikace dabrafenibu a užití v multiterapii
Je nutno zmínit, že dabrafenib bude provázet podobný problém jako konkurenční inhibitor vemurafenib – pro zamezení podávání skupinám pacientů, na něž nemá nejmenší pozitivní účinek, bude nutno zavést genetické testování nádorové tkáně na přítomnost aktivačních mutací kinázy B-RAF, konkrétně na mutace v aminokyselině B-RAFV600. Za tímto účelem je samozřejmě zcela nepostačující testování somatických mutací, bude nutno vyžadovat kvalifikované testování vzorku DNA získaného přímo z nádorové tkáně (alternativou může v budoucnu být nově vyvíjené testování fragmentů cirkulující nádorové DNA zvané cfDNA, ze které lze pomocí vysoce senzitivních metod detekovat přítomnost příslušné mutace [30]). Lék nejlépe funguje proti nádorům s mutací B-RAFV600E, ale je schopen se s menší intenzitou vázat i na cílovou molekulu nesoucí dvě méně často se vyskytující záměny B-RAFV600K a B-RAFV600D (je pravděpodobné, že zejména u mutace B-RAFV600D bude vhodné předepisovat vyšší dávkování). Podávání léčiva pacientům s nádory exprimujícími pouze běžnou nemutovanou kinázu B-RAF není vhodné z důvodu neúčinnosti. Vhodné není ani podávání léčiva pacientům nesoucím jiné mutace kinázy B-RAF a pravděpodobně ani těm, u kterých by byly zjištěny mutace v proteinech rodiny RAS.
Zde je nutno zastavit se u fenoménu opakovaně asociovaného s inhibitory kinázy B-RAF, a to se vznikem de novo mutací v MAP kinázové dráze následkem podávání inhibitorů B-RAF kinázy. Tyto de novo vznikající mutace zcela eliminují pozitivní působení podávaného léčiva, a naopak jsou dokonce schopny způsobit, že podávání léčiva urychluje růst nádoru. Ačkoliv se na jejich výskyt u dostatečně velkého množství pacientů užívajících dabrafenib zatím nikdo nezaměřil, můžeme si průběh vzniku těchto mutací ilustrovat na dnes již velmi dobře popsaných případech známých z počátků podávání jiného inhibitoru kinázy B-RAF, vemurafenibu. Podávání inhibitoru vede sice k inaktivaci kinázy B-RAF, ale nepotlačuje (nýbrž dokonce podporuje) její translokaci na buněčnou membránu. Kinázy rodiny RAF působí na membráně jako dimery, přičemž ony dvě molekuly v dimeru jsou schopny navzájem stimulovat svou aktivitu. Dimery mohou sestávat jak pouze z kinázy B-RAF (tj. homodimery), tak i z jiných RAF kináz (např. heterodimer B-RAF a C-RAF). V případě heterodimerů patrně postačuje i samotná přítomnost neaktivní kinázy B-RAF ke stimulaci jejího vazebného partnera, tj. kinázy C-RAF, a k další signalizaci prostřednictvím MAP kinázové dráhy. Výsledný signál je samozřejmě o to slabší, o co více vazebných partnerů je reprezentováno kinázou B-RAF a o co méně se na tvorbě homo - a heterodimerů podílí kináza C-RAF. Až potud jde o normální fyziologický jev, který při léčbě nádoru ničemu nevadí. Jenže aktivace rodiny RAF kináz je spouštěna prostřednictvím G proteinu RAS. Pokud se tento protein stane hyperaktivním pomocí de novo vzniklé mutace (což se v nádoru léčeném inhibitory kinázy B-RAF stává nepříjemně často), pak samotná signalizace přes C-RAF je plně postačující pro stimulaci celé MAP kinázové dráhy. Pokud k tomu přidáme ještě nepříjemnou vlastnost inhibitorů kinázy B-RAF spočívající ve stimulaci tvorby navzájem se transaktivujících heterodimerů kináz B-RAF a C-RAF na plazmatické membráně, je zaděláno na vážný problém. Látka, která měla původně růst nádoru potlačovat (vemurafenib a pravděpodobně i dabrafenib), jej v kombinaci s mutací v G proteinu RAS je schopna dokonce stimulovat. Může tak docházet nejen k obnovení progrese samotného melanomu; identické mutace bývají typické i pro maligní nádory plic [2,31,32].
Progresi nemoci při podávání dabrafenibu významně ovlivňuje přítomnost mutací či delecí ve fosfatáze PTEN. U pacientů s maligním melanomem a záměnou B-RAFV600E či B-RAFV600K došlo ke zkrácení průměrné doby bez progrese nemoci z 6,9 na 4,2 měsíce v případě přítomnosti nesprávně fungující fosfatázy PTEN [33]. Mutací ovlivňujících účinnost léčby může být víc, u vemurafenibu je mimo jiné znám opakovaný vznik rezistence na léčbu následkem tvorby mutací v kináze PDGFRβ [34]. U dabrafenibu existují zatím jen data ze studií na buněčné úrovni, kdy bylo zjištěno, že klony rezistentní k dabrafenibu (i vemurafenibu) nesou delece v MEK1 či N-RAS [35,36].
Výrobce dabrafenibu zjevně předpokládá, že preparát bude předepisován především v kombinaci s jinými léčivy pro zamezení četných relapsů nemoci pozorovaných u všech dosud vyvinutých a formou monoterapie podávaných inhibitorů kinázy B-RAF. Kombinovaná terapie je výhodná i s ohledem na to, že samotná aktivační mutace v kináze B-RAF způsobuje pouze vznik benigních névů [37]. GlaxoSmithKline má nyní v řízení čtyři patentové přihlášky pro kombinaci dabrafenibu s inhibitorem kinázy AKT na bázi furankarboxamidu (WO-2011044414) nebo thiophenekarboxamidu (WO-2011044415). Další patentové přihlášky se zaměřují na kombinaci dabrafenibu s inhibitorem PI3K/mTOR dráhy prostřednictvím látky GSK-2126458 (WO-2011046894), popřípadě s trametinibem inhibujícím kinázu MEK (WO-2011047238) [14]. Nutno však dodat, že dle prvních výsledků klinických studií zaměřených na podávání dabrafenibu v kombinaci s trametinibem dochází i zde ke kompletní odpovědi jen vzácně [29]. Zdá se však, že ke zlepšení přežití při užití této kombinace dochází, dobře experimentálně dokumentovanou výhodou je i snížené množství vedlejších účinků [18,29].
Příběh dabrafenibu a vemurafenibu jasně naznačuje budoucnost biologické léčby: bude nutné testování nádorových biopsií odebíraných v různých stadiích léčby pro úpravu spektra podávaných léčiv v odpovědi na nově vznikající mutace, jimiž se nádor doslova přizpůsobuje ztíženým podmínkám pro svůj růst. Tato praxe se výrazně liší od praxe dnešní, kdy řada zejména menších českých onkologických pracovišť nevyužívá možností současné diagnostiky nad rámec povinností stanovených vyhláškou a kdy tedy opakovaně dochází k nadužívání biologické léčby tam, kde je sice v souladu s legislativou, avšak její dopad na pacienta je v lepším případě nulový, častěji však dokonce negativní (žádný účinek na růst nádoru, avšak velký potenciál vzniku vedlejších účinků). S tím, jak se jednotlivá biologická léčiva dostávají i na český trh, bude velmi žádoucí zpřísnit (popřípadě zcela nově stanovit) pravidla, jimiž se podávání těchto biologických léčiv řídí, aby byla v souladu se současným stavem poznání klinické vědy.
Práce byla podpořena projekty Univerzity Karlovy v Praze UNCE 204015 a PRVOUK P31/2012, projektem Grantové agentury České republiky P301/12/1686 a projektem Interní grantové agentury Ministerstva zdravotnictví České republiky NT13663-3/2012.
Autor deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
RNDr. Petr Heneberg, Ph.D.
3. LF UK v Praze
Ruská 87
100 00 Praha 10
e-mail: petr.heneberg@if3.cuni.cz
Obdrženo: 29. 5. 2012
Přijato: 22. 8. 2012
Zdroje
1. Jailkhani N, Chaudhri VK, Rao KV. Regulatory cascades of protein phosphatases: implications for cancer treatment. Anticancer Agents Med Chem 2011; 11(1): 64–77.
2. Heneberg P. Pokroky v klinické léčbě zhoubného melanomu: inhibice kinázy B-RAF. Klin Onkol 2011; 24(4): 256–264.
3. Ferlay J, Shin HR, Bray F et al. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10 [online]. International Agency for Research on Cancer, Lyon, Francie; c2010 [aktualizováno 23. května 2012; citováno 23. května 2012]. Available from: http://globocan.iarc.fr.
4. Middleton MR, Lorigan P, Owen J et al. A randomized phase III study comparing dacarbazine, BCNU, cisplatin and tamoxifen with dacarbazine and interferon in advanced melanoma. Br J Cancer 2000; 82(6): 1158–1162.
5. Chapman PB, Einhorn LH, Meyers ML et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 1999; 17(9): 2745–2751.
6. Balch CA, Atkins MB, Sober AJ. Cutaneous melanoma. In: DeVita VT Jr, Hellman S, Rosenberg SA (eds). Cancer, principles and practice of oncology. 7th ed. Philadelphia: Lippincott Williams & Wilkins 2005.
7. Hodis R, Watson IR, Kryukov GV et al. A landscape of driver mutations in melanoma. Cell 2012; 150(2): 251–263.
8. Houben R, Terheyden P, Bröecker EB et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog 2004; 3(1): 6.
9. Long GV, Menzies AM, Nagrial AM et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 2011; 29(10): 1239–1246.
10. Chapman PB, Hauschild A, Robert C et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364(26): 2507–2516.
11. Chapman PB, Hauschild A, Robert C et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAFV600E-mutated melanoma. J Clin Oncol 2012; 30: Abstrakt 8502.
12. Vyzula R, Adámková Krákorová D, Babjuk M et al. Zásady cytostatické léčby maligních onkologických onemocnění. 14th ed. Brno: Masarykův onkologický ústav 2012.
13. Šebek V. Léčivý přípravek Zelboraf byl registrován v EU pro léčbu pacientů s agresivní formou melanomu. Klin Onkol 2012; 25(2): 143–144.
14. Heneberg P. Dabrafenib, a small-molecule oral inhibitor of the mutant kinase BRAF for the potential treatment of cancer, in particular melanoma [online]. Thomson Reuters Pharma and Thomson Reuters Pharma Partnering databases; [aktualizováno 23. března 2012; citováno 23. března 2012]. Available from: https://partnering.thomson-pharma.com/partnering/partnering/.
15. Adams JL, Dickerson SH, Johnson NW et al. Benzene sulfonamide thiazole and oxazole compounds. International Patent claim WO/2009/137391 (podaný 6. 5. 2008), schválen v USA jako US-07994185, platný do ledna 2030.
16. King AJ, Arnone M, Moss K et al. A selective Raf Kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-Raf V600E mutation. San Francisco: 21st AACR-NCI-EORTC Int Congress, 17. 11. 2009: Abstrakt B88.
17. Ouellet D, Laquerre S, Minthorn E et al. Predictions of therapeutic doses of a B-Raf inhibitor, GSK2118436, based on exposure-response modeling of preclinical tumor biomarker and xenograft data. Orlando: 102nd Am Assoc Cancer Res Ann Meet, 6. 4. 2011: Abstrakt 5035.
18. Infante J, Falchook G, Lawrence D et al. Phase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK-1120212 dosed in combination with the oral BRAF inhibitor GSK-2118436. Chicago: 47th Am Soc Clin Oncol Ann Meet, 6. 6. 2011: Abstrakt CRA8503.
19. GlaxoSmithKline PLC. Study BRF113468: An Open Label Study to Examine the Effects of a High-Fat Meal and Particle Size on the Pharmacokinetics of Orally Administered GSK2118436 in Subjects with BRAF Mutation-Positive Tumor. GSK Clinical Study Register 2012.
20. Kefford R, Arkenau H, Brown MP. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. Chicago: 46th Am Soc Clin Oncol Ann Meet, 5. 6. 2010: Abstrakt 8503.
21. GlaxoSmithKline PLC. Study BRF112680: A Phase I, Open-Label, Multiple-Dose, Dose-Escalation Study to Investigate the Safety, Pharmacokinetics, and Pharmacodynamics of the BRAF Inhibitor GSK2118436 in Subjects with Solid Tumors. GSK Clin Study Register 2012.
22. Azer MW, Menzies AM, Haydu L et al. Patterns of progression in patients (pts) with V600 BRAF-mutated melanoma metastatic to the brain treated with dabrafenib (GSK2118436). J Clin Oncol 2012; 30: Abstrakt 8558.
23. Kirkwood JM, Long GV, Trefzer U et al. BREAK-MB: A phase II study assessing overall intracranial response rate (OIRR) to dabrafenib (GSK2118436) in patients (pts) with BRAF V600E/K mutation-positive melanoma with brain metastases (mets). J Clin Oncol 2012; 30: Abstrakt 8501.
24. Prahallad A, Sun C, Huang S et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483(7387): 100–103.
25. Corcoran RB, Falchook GS, Infente JR et al. BRAF V600 mutant colorectal cancer (CRC) expansion cohort from the phase I/II clinical trial of BRAF inhibitor dabrafenib (GSK2118436) plus MEK inhibitor trametinib (GSK1120212). J Clin Oncol 2012; 30: Abstrakt 3528.
26. Hauschild A, Grob JJ, Demidov LV et al. Phase III, randomized, open-label, multicenter trial (BREAK-3) comparing the BRAF kinase inhibitor dabrafenib (GSK2118436) with dacarbazine (DTIC) in patients with BRAFV600E-mutated melanoma. J Clin Oncol 2012; 30: Abstrakt LBA8500.
27. Kefford R. Selective inhibition of oncogenic BRAF V600E/K/D by GSK2118436: Evidence of clinical activity in subjects with metastatic melanoma. Florida: 7th Int Melanoma Res Congress, 5. 11. 2010.
28. Anforth R, Blumetti T, Kefford R et al. Cutaneous manifestations of the selective BRAF inhibitor GSK2118436 phase I/II. Australas J Dermatol 2011; 52 : 10.
29. Weber JS, Flaherty KT, Infante JR et al. Updated safety and efficacy results from a phase I/II study of the oral BRAF inhibitor dabrafenib (GSK2118436) combined with the oral MEK 1/2 inhibitor trametinib (GSK1120212) in patients with BRAFi-naive metastatic melanoma. J Clin Oncol 2012; 30: Abstrakt 8510.
30. Long GV, Ascierto PA, Grob JJ et al. Tumor-specific circulating cell-free DNA (cfDNA) BRAF mutations (muts) to predict clinical outcome in patients (pts) treated with the BRAF inhibitor dabrafenib (GSK2118436). J Clin Oncol 2012; 30: Abstrakt 8518.
31. Nazarian R, Shi H, Wang Q et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468(7326): 973–977.
32. Cichowski K, Jänne PA. Drug discovery: inhibitors that activate. Nature 2010; 464(7287): 358–359.
33. Nathanson KL, Martin A, Letrero R et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor GSK-2118436 (GSK-436). Chicago: 47th Am Soc Clin Oncol Ann Meet, 6. 6. 2011: Abstrakt 8501.
34. Villanueva J, Vultur A, Lee JT et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinases switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18(6): 683–695.
35. Ascierto PA, Gentilcore G, Madonna G et al. Are GSK2118436 and GSK1120212 effective in melanoma cell lines harboring V600BRAF mutations different from the common V600EBRAF variant? Eur J Cancer 2011; 47: S11.
36. Greger JG Jr, Eastman SD, Zhang V et al. Acquired resistance to the BRAF inhibitor GSK2118436, mediated by NRAS or MEK mutation, can be overcome with combinations that inhibit BRAF and MEK, BRAF and PI3K/mTOR, or MEK and PI3K/mTOR. Florida: 23rd AACR-NCI-EORTC Int Congress, 14. 11. 2011: Abstrakt B71.
37. Michaloglou C, Vredeveld LC, Soengas MS et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436(7051): 720–724.
38. Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer 2011; 104(3): 392–398.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2012 Číslo 5
- Brno opět přivítá onkology a nelékařské zdravotnické pracovníky
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Fixní kombinace tramadol/paracetamol je doporučenou volbou v léčbě chronické bolesti v ordinaci praktického lékaře
Najčítanejšie v tomto čísle
- Prognostické faktory konvenčního osteosarkomu dospělých pacientů
- Sarkómy maternice – prehľad
- Septické komplikace intravenózních portových systémů – kazuistiky a přehled literatury
- Mutace BRAF: nový přístup k cílené léčbě melanomu