
Stanovení nabumetonu a kyseliny 6‑methoxy-2-naftyloctové v plazmě pomocí HPLC s UV a MS detekcí
Determination of nabumetone and 6-methoxy-2-naphthylacetic acid in plasma using HPLC with UV and MS detection
The study aimed to establish and validate an analytical method for the determination of nabumetone and 6-methoxy-2-naphthylacetic acid (6-MNA) in human plasma after a single therapeutic dose of the drug. Two methods based on HPLC with UV and MS detection were compared. Optimal results in sample preparation were achieved using solid phase extraction. The recovery reached approximately 84% and 86–90% for nabumetone and 6-MNA, respectively. A reverse C18 column was used for HPLC separation of the analytes. The limit of UV detection was 50 nM and 0.1 μM for 6-MNA and nabumetone, respectively. The limit of MS detection was 1 μM and 0.5 μM for 6‑MNA and nabumetone, respectively. Precision ranged between 4.2–14.4% and 4.6–8.5% using UV and MS detection for nabumetone, respectively. The respective values for 6-MNA were 2.4–12.5% and 2.1–9.4%. Accuracy ranged between 93.4–109.6% in UV detection and 86.2–107.9% using UV and MS detection for nabumetone, respectively. The respective values for 6-MNA were 87.8–107.4% and 86.3–106.4%. The method was subsequently applied to determine the pharmacokinetic parameters of nabumetone and 6-MNA in a group of 24 healthy volunteers.
Key words:
nabumetone – pharmacokinetics – 6-methoxy-2-naphthylacetic acid – HPLC – LC/MS
Autoři:
Lenka Nespěšná 1,2; Martin Štícha 1; Olga Matoušková 2; František Perlík 2; Ondřej Slanař 2
Působiště autorů:
Univerzita Karlova v Praze, Katedra organické a jaderné chemie, Přírodovědecká fakulta
1; Univerzita Karlova v Praze, Farmakologický ústav 1. lékařské fakulty
2
Vyšlo v časopise:
Čes. slov. Farm., 2011; 60, 17-24
Kategorie:
Původní práce
Souhrn
Cílem práce bylo zavést a validovat analytickou metodu, která by umožnila detekovat koncentrace nabumetonu a kyseliny 6-methoxy-2-naftyloctové (6-MNA) po jednorázovém podání terapeutické dávky léčiva. Byly porovnány dvě metody stanovení pomocí HPLC s UV a hmotnostní detekcí. Při úpravě vzorku bylo optimálních výsledků dosaženo pomocí extrakce na pevné fázi (SPE). Výtěžnost se pohybovala okolo 84 % pro nabumeton a 86–90 % pro 6-MNA. HPLC separace analytů byla prováděna na reverzní C18 koloně. Limit UV detekce pro 6-MNA byl 50 nM, pro nabumeton 0,1 μM. Limit MS detekce pro 6-MNA činil 1 μM, pro nabumeton 0,5 μM. Přesnost metody pro stanovení nabumetonu se pohybovala v rozmezí 4,2–14,4 % při UV detekci a 4,6–8,5 % při MS detekci. Přesnost metody pro 6-MNA byla 2,4–12,5% při UV detekci a 2,1–9,4 % při MS detekci. Správnost metody pro stanovení nabumetonu se pohybovala v rozmezí 93,4–109,6 % při UV detekci a 86,2–107,9 % při MS detekci. Správnost metody pro 6-MNA byla 87,8–107,4 % při UV detekci a 86,3–106,4 % při MS detekci. Vhodnost metody byla ověřená na vzorcích pro stanovení farmakokinetiky léčiva u 24 zdravých dobrovolníků.
Klíčová slova:
nabumeton – farmakokinetika – kyselina 6-methoxy-2-naftyloctová – HPLC – LC/MS
Úvod
Nabumeton (4-(6-methoxy-2-naftyl)-butan-2-on) patří mezi nesteroidní protizánětlivé látky vykazující preferenční inhibici cyklooxygenázy 2 1). Je používán v léčbě akutních i chronických symptomů revmatoidní artritidy a osteoartrózy 2, 3). Nabumeton je proléčivo, které nevykazuje protizánětlivou aktivitu, zatímco jeho hlavní metabolit kyselina 6-methoxy-2-naftyloctová (6-MNA) vykazuje farmakodynamické účinky antiflogistické, antipyretické a analgetické.
Přestože jsou klinické účinky nabumetonu dobře popsány, farmakokinetika nabumetonu dosud podrobněji popsána nebyla kvůli příliš nízkým koncentracím mateřské látky v krvi po podání terapeutických dávek. Z důvodu nízkých hladin nabumetonu v krvi je dosud publikována jen jedna metoda HPLC s fluorimetrickou detekcí nabumetonu pro použití v klinických studiích 4). Dobře je naopak popsána farmakokinetika 6-MNA díky dostupnosti dostatečně citlivých analytických technik 5–9).
Cílem naší práce bylo zavést a validovat analytickou metodu, která by umožnila detekovat koncentrace nabumetonu a 6-MNA po jednorázovém podání terapeutické dávky léčiva.
Pokusná část
Chemikálie
Nabumeton p.a. (Sigma-Aldrich), 6-MNA p.a. (MP Biomedicals), naproxen 98% (Sigma-Aldrich), methanol pro HPLC (Lach-Ner), aceton p.a. (Merck), propan-2-ol p.a. (Lach-Ner) kyselina o-fosforečná 85% (Merck), fosforečnan sodný terciární čistý (Lachema), acetonitril pro HPLC (Lach-Ner), kyselina trifluorooctová 99% (Sigma-Aldrich), deionizovaná voda (Millipore), dusík 4.6 (Linde Gas), helium 4.6 (Linde Gas).
Příprava vzorků
Vzorky krve jsme odebírali do zkumavek Vacutainer s přídavkem heparinu. Ihned po odebrání krve jsme oddělili plasmu centrifugací při 3000 g. Plasma byla poté uložena do -70 °C až do doby analýzy.
Pro předúpravu vzorků jsme zvolili techniku extrakce na tuhou fázi (SPE). K 0,5 ml vzorku jsme přidali 50 μl roztoku vnitřního standardu o koncentraci 0,1 mmol/l. Vzorek jsme naředili 0,5 ml fosfátového pufru pH 2 (10 mM), okyselili 50 μl 10% kyseliny fosforečné a řádně promíchali.
Takto připravený vzorek jsme aplikovali na kolonku Discovery® DSC-18 (1 ml, 100 mg), Supelco, USA předem promytou 2 ml methanolu a 2 ml fosfátového pufru pH 2 (10 mM). Všechny roztoky protekly rychlostí 1 ml.min-1.
Následně jsme kolonku prosávali vzduchem po dobu 5 minut. Zachycené látky byly vymyty 2 ml methanolu. Získaný extrakt jsme odpařili dosucha při 45 °C pod mírným proudem dusíku. Reziduum jsme poté rozpustili ve 250 μl mobilní fáze a 30 μl nastříkli do HPLC.
HPLC/UV/MS analýza
Pro analýzu extraktu jsme použili kapalinový chromatograf HP 1100 s UV DAD, Hewlett Packard, USA. Jako hmotnostní detektor jsme použili přístroj Esquire 3000, Bruker Daltonics, Německo.
Separace probíhala na koloně Supelcosil LC-18 15 cm × 4,6 mm 5 μm, Supelco, USA a jako mobilní fázi jsme použili směs acetonitrilu a 0,1% TFA v poměru 50 : 50 (v/v) o průtokové rychlosti 0,3 ml.min-1.
UV detekce proběhla při vlnové délce absorpčního maxima sledovaných látek λ = 230 nm. Současně jsme sledovali i hmotnostní detekci s chemickou ionizací za atmosférického tlaku v pozitivním módu. Podmínky APCI detekce byly následující: rozsah skenu 50–800 u, teplota sušícího plynu 250 °C, teplota iontového zdroje 400 °C, tlak plynu v zamlžovači 30 psi, průtok sušícího plynu (N2) 5 l.min-1, napětí na kapiláře 4 kV.
Vyhodnocení jsme provedli podle chromatogramu náležejícího nejintenzivnějšímu iontu ve spektru příslušné látky. Pro 6-MNA a nabumeton to byl ion m/z 171 a pro vnitřní standard – naproxen ion m/z 185.
Validace metody
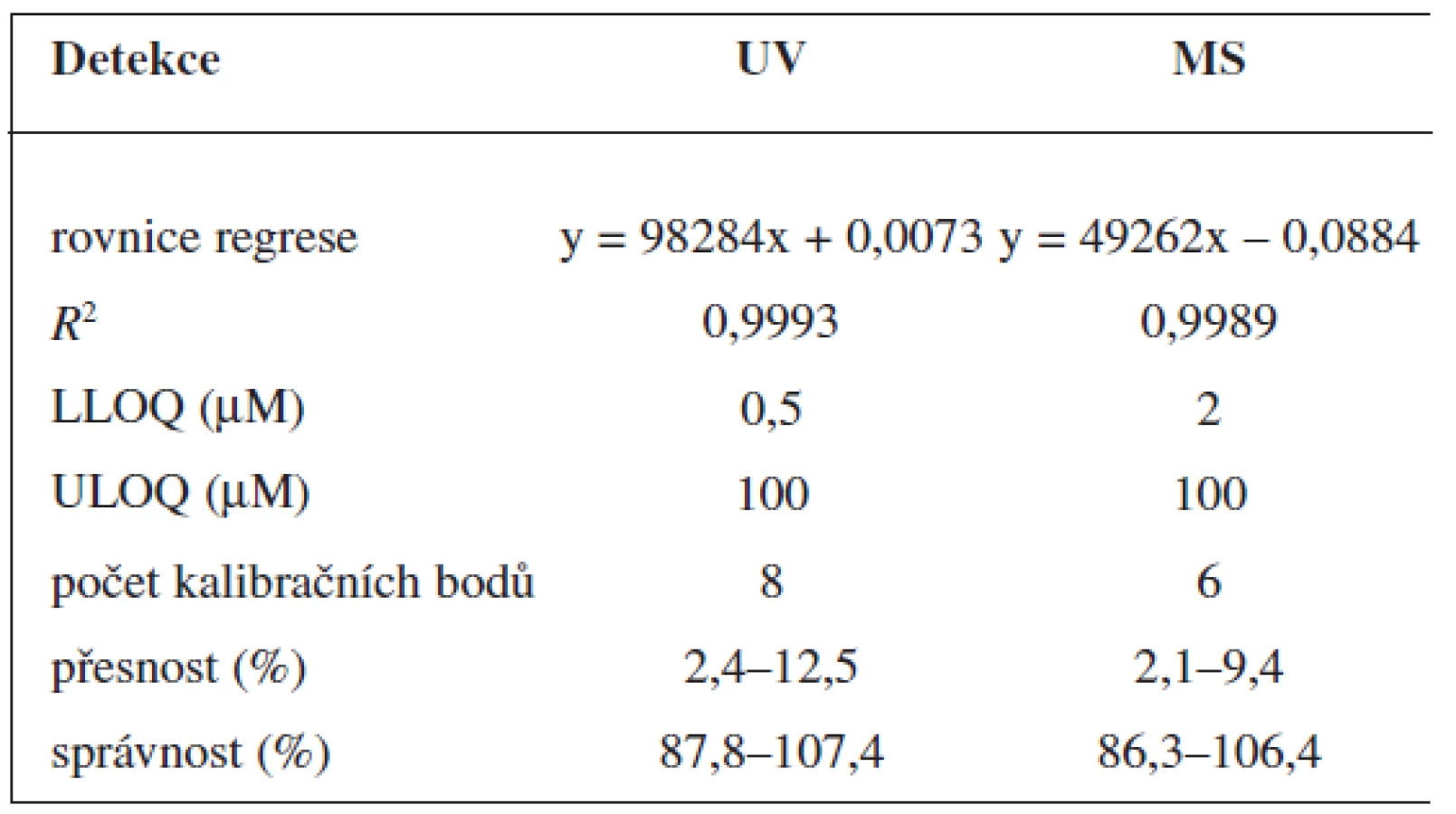
Validaci metody jsme provedli na základě požadavků pro validaci bioanalytických metod 10). V souladu s těmito požadavky jsme stanovili mez stanovitelnosti (LLOQ), správnost, přesnost, výtěžnost, linearitu a selektivitu detekce.
Pro nabumeton i 6-MNA byly sestrojeny kalibrační křivky. Vzorky o známé koncentraci s přídavkem vnitřního standardu byly zpracovány SPE a dále analyzovány pomocí HPLC s UV a MS detekcí. Pro 6-MNA byly připraveny roztoky o těchto koncentracích: 0,2; 0,5; 1; 2; 5; 10; 20; 50 a 100 μmol/l pro nabumeton 0,2; 0,5; 1; 2; 5; 10 a 20 μmol/l.
Mez stanovitelnosti jsme určili jako nejnižší bod kalibrační křivky, kde pík analytu je indentifikovatelný, oddělený a reprodukovatelný s přesností 20 % a správností 80–120 %.
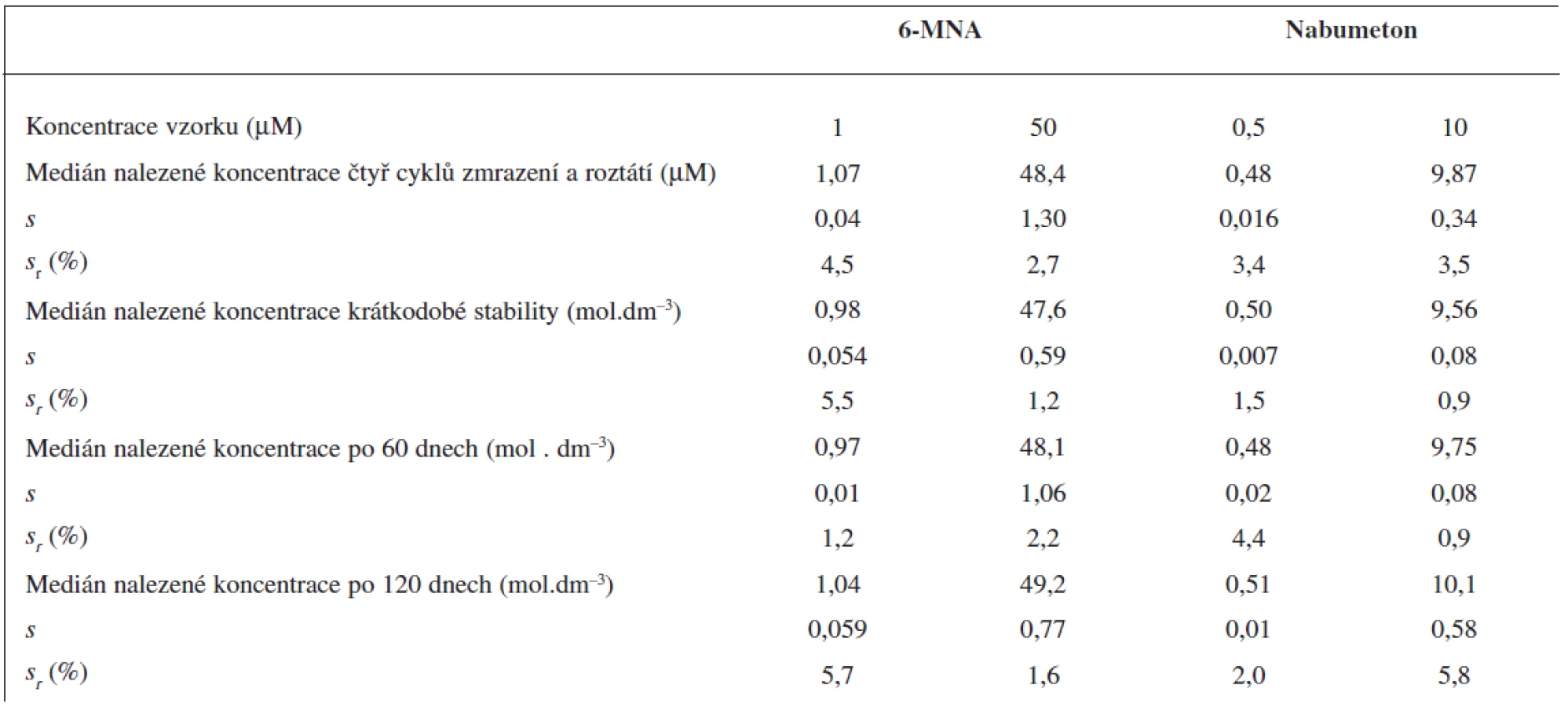
Součástí validace metody bylo také studium stability zásobních roztoků a vzorků. V případě zásobních roztoků analytů a vnitřního standardu jsme měřili koncentrace látek přítomných ve vzorku v závislosti na čase u skladovaných roztoků po nástřiku 5 μl zásobního roztoku. Detekční podmínky byly shodné s podmínkymi při měření extraktů z plazmy. První měření proběhlo ihned po jejich přípravě.
Dále jsme sledovali vliv zmrazení a roztátí vzorků v několika cyklech na koncentraci analytů ve vzorku. Vzorky o vysoké a nízké koncentraci analytů (množství dostatečné pro tři analýzy na koncentraci, pro 6-MNA 1 a 50 μM; pro nabumeton 0,5 a 10 μM) jsme skladovali při teplotě -70 °C po dobu 24 hodin. Poté roztály při laboratorní teplotě a následně byly znovu zmraženy. Tento postup jsme opakovali a po čtvrtém cyklu roztátí vzorků jsme provedli analýzu.
Pro studium krátkodobé teplotní stability jsme připravili vzorky o vysoké a nízké koncentraci (pro 6-MNA 1 a 50 μM; pro nabumeton 0,5 a 10 μM, tři alikvoty od každé). Vzorky byly ponechány při laboratorní teplotě 6 hodin a následně zpracovány.
Pro stanovení dlouhodobé teplotní stability vzorků jsme připravili vzorky o vysoké a nízké koncentraci analytů (pro 6-MNA 1 a 50 μM; pro nabumeton 0,5 a 10 μM). Vzorky jsme uchovávali při teplotě -70 °C a v časových intervalech je analyzovali ve třech stanoveních v každé koncentraci.
Reprodukovatelnost metody jsme zhodnotili na základě výsledků analýz kontrolních vzorků, které jsme provedli v průběhu pěti týdnů. Celkem jsme analyzovali deset sérií po třech koncentracích (nízké, střední a vysoké) pro oba analyty. 6-MNA byla sledována na koncentračních úrovních 1; 10 a 50 μM, nabumeton pak v koncentracích 0,5; 2 a 10 μM.
Aplikace metody
Metodu jsme následně použili na stanovení farmakokinetických parametrů nabumetonu a 6-MNA u 24 zdravých dobrovolníků po podání 500 mg nabumetonu (Relifex, MEDA AB Švédsko). Zdraví dobrovolníci byli po vstupním fyzikálním, biochemickém a hematologickém vyšetření krátkodobě hospitalizováni na našem oddělení. Během hospitalizace dostal každý dobrovolník jednorázovou dávku přípravku Relifex s 250 ml vody na zapití. Podání léčiva probíhalo vždy v ranních hodinách pod dohledem zdravotní sestry. Po dobu studie měli dobrovolníci volný přístup k pitné vodě ad libitum. První lehké standardizované jídlo bylo podáváno za 4 hodiny od podání nabumetonu. Odběry jsme prováděli v čase před podáním léčiva a 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72 a 96 hodin po podání léku. Tato studie byla schválena Etickou komisí VFN a všichni dobrovolníci podepsali informovaný souhlas před zařazením do studie.
Výsledky a diskuze
Optimalizace metody
HPLC separace analytů proběhla na reverzní C18 koloně. Testovali jsme mobilní fáze o složení acetonitril/voda a acetonitril/0,1% TFA. Chromatografická separace byla ve všech případech provedena isokratickou eluční technikou.
Jako výchozí bod jsme zvolili mobilní fázi acetonitril : voda 50 : 50 (v/v), kdy jsme pozorovali dobrou separaci analytů a vnitřního standardu. Zjistili jsme, že rostoucí průtoková rychlost měla negativní vliv na symetrii píků a odezva MS detekce (výška píku) při překročení průtoku 0,3 ml.min-1 klesala.
Při použití mobilní fáze acetonitril: 0,1% TFA v poměru 50 : 50 (v/v) došlo k očekávanému vylepšení symetrie píků a k navýšení signálu MS, nicméně při překročení průtokové rychlosti 0,3 ml.min-1 i za těchto podmínek účinnost ionizace MS klesala.
Vyšší obsah acetonitrilu vedl ke snížení retence a zhoršení symetrie píků. Odezva MS při obsahu acetonitrilu 50 % a více setrvávala na přibližně konstantní úrovni. Pro další práci jsme zvolili poměr acetonitril : 0,1% TFA 50 : 50 (v/v).
MS detekce
Současné požadavky na validaci bioanalytických metod vyžadují průkaz nepřítomnosti matričních efektů. V naší práci jsme tento průkaz nezjišťovali, protože jsme použili metodu chemické ionizace za atmosferického tlaku (APCI), která je podstatně robustnější vzhledem ke složení matrice a riziko matričních efektů je významně nižší ve srovnání s metodou elektrosprejové ionizace (ESI). Pro použití v rámci TDM, ke kterému jsme naší metodu vyvinuli, lze proto popisovanou metodu považovat za dostatečně robustní s ohledem na matriční efekty.
Při použití chemické ionizace za atmosferického tlaku všechny tři sledované látky poskytovaly signál pouze při záznamu kladných iontů.
Na obrázku 1 je zobrazeno spektrum 6-MNA, kde lze vidět ion m/z 217, který je tvořen protonovanou molekulou (M+H)+ a fragmentový ion m/z 171, vzniklý dekarboxylací molekuly. Ion m/z 282 nepatří sledované látce, jedná se o ion z pozadí. Na obrázku 2 je APCI spektrum naproxenu. Opět je zde pozorovatelná protonovaná molekula (M+H)+ o hmotě m/z 231 a intenzivní fragmentový ion m/z 185 vzniklý dekarboxylací.


Na obrázku 3 je zobrazeno spektrum nabumetonu. Ion m/z 229 patří protonované molekule (M+H)+ a intenzivní m/z 171 je fragment vzniklý odštěpením části postranního řetězce (M–CH3COCH2)+. Ion m/z 211 pak odpovídá ztrátě 17 u. Ostatní přítomné ionty patří k signálu pozadí.

Validace
Metoda stanovení 6-MNA a nabumetonu splňuje požadavky na přesnost a správnost. Selektivita metody byla testována analýzou deseti „slepých“ vzorků, přičemž jsme nepozorovali žádné interference.
Zároveň jsme určili detekční limity pro obě stanovované látky. Limit UV detekce pro 6-MNA byl 50 nM, pro nabumeton 0,1 μM. Limit MS detekce pro 6-MNA činil 1 μM, pro nabumeton 0,5 μM.
V přítomnosti naproxenu jako vnitřního standardu (10 μM) byla v koncentračním rozmezí 0,2–200 μM na deseti koncentračních úrovních proměřena odezva standardních roztoků 6-MNA a nabumetonu s využitím UV detekce. Rozmezí pro MS detekci bylo 0,5–200 μM, devět koncentračních úrovní.
Odezva byla pro oba analyty v uváděném rozsahu lineární při použití UV i MS detekce (obr. 4 až 7).




Validace byla provedena pro UV a pro MS detekci zvlášť. Parametry validace pro stanovení 6-MNA jsou uvedeny v tabulce 1. Validace metody stanovení nabumetonu je shrnuta v tabulce 2. Výtěžnost se pohybovala okolo 84 % pro nabumeton a 86–90 % pro 6-MNA (tab. 3).


Stabilita zásobních roztoků
Stabilitu zásobních roztoků jsme sledovali v intervalech 14, 30, 60 a 120 dní. V žádném z intervalů se plochy píku pro 6-MNA, nabumeton nebo vnitřní standard neodchylovaly o více než 2 % od bazálních hodnot.
Stabilita vzorků krevní plazmy
Čtyři cykly zmrazení a opětovného roztátí nevedly k významnému poklesu obsahu analytů ve vzorku.
Krátkodobou teplotní stabilitu jsme sledovali u vzorků krevní plazmy, kdy jsme roztoky ponechali při laboratorní teplotě po dobu 6 hodin. Po této době opět nedošlo k významnému poklesu obsahu analytů ve vzorku podobně jako při testech dlouhodobé stability po 60 a 120 dnech skladování při -70 °C. Všechny výsledky stabilit testů krevní plazmy jsou sumarizovány v tabulce 4.
Reprodukovatelnost metody
Relativní směrodatná odchylka stanovení 6-MNA v kontrolních vzorcích na třech různých koncentračních hladinách se pohybovala v rozmezí 2,9–6,1 %. Obdobně nabývala i v případě nabumetonu relativní směrodatná odchylka hodnot 1,6–3,6 %.
Aplikace metody
Průměrné hladiny (95% CI) nabumetonu a 6-MNA získané UV detekcí jsou uvedeny na obrázcích 8 a 9. Základní farmakokinetické parametry nabumetonu a 6‑MNA jsou uvedeny v tabulce 5.



Závěr
Při srovnání obou detekčních technik, lze říci, že UV detekce, jak dokládají směrnice z rovnic kalibračních přímek, je stejně citlivá pro 6-MNA i pro nabumeton. Pokud stejným způsobem zhodnotíme hmotnostní detekci, je citlivější pro nabumeton (směrnice kalibrační přímky nabumetonu je přibližně 2× než směrnice kalibrační přímky 6-MNA). Při použití UV detekce jsme u obou analytů dosáhli nižší meze stanovitelnosti než v případě detekce hmotnostní. Lze předpokládat, že použitím techniky SIM u kvadrupólového hmotnostního spektrometru, případně techniky MRM u tandemových přístrojů by bylo možné zvýšit citlivost o několik řádů a dostat se minimálně na úroveň UV detekce. Tyto módy detekce však námi použité přístrojové vybavení neumožnilo.
Zavedené metody UV i hmotnostní detekce umožňují stanovit koncentrace nabumetonu a 6-MNA po jednorázovém podání terapeutické dávky léčiva.
Práce vznikla za podpory grantu GA UK 52707.
Došlo 26. listopadu 2010
Přijato 20. prosince 2010
Adresa pro korespondenci:
doc. MUDr. Ondřej Slanař, Ph.D.
Farmakologický ústav 1. LF UK
Albertov 4, 128 00 Praha 2
e-mail: oslan@lf1.cuni.cz
Zdroje
1. Boyle, E. A., Freeman, P. C., Mangan, F. R., Thomson, M. J.: Nabumetone (BRL 14777, 4-[6-methoxy-2-naphthyl]-butan-2-one): a new anti-inflammatory agent. J Pharm Pharmacol. 1982; 34, 562–569.
2. Battisti, W. P., Katz, N. P., Weaver, A. L., Matsumoto, A. K., Kivitz, A. J., Polis, A. B., Geba, G. P.: Pain management in osteoarthritis: a focus on onset of efficacy – a comparison of rofecoxib, celecoxib, acetaminophen, and nabumetone across four clinical trials. J Pain. 2004; 5, 511–520.
3. Hedner, T., Samulesson, O., Wahrborg, P., Wadenvik, H., Ung K. A., Ekbom, A.: Nabumetone: therapeutic use and safety profile in the management of osteoarthritis and rheumatoid arthritis. Drugs. 2004; 64, 2315–2343; discussion 2344–2315.
4. Kobylinska, K., Barlinska, M., Kobylinska, M.: Analysis of nabumetone in human plasma by HPLC. Application to single dose pharmacokinetic studies. J Pharm Biomed Anal. 2003; 32, 323–328.
5. Starek, M., Krzek, J.: A review of analytical techniques for determination of oxicams, nimesulide and nabumetone. Talanta. 2009; 77, 925–942.
6. Kendall, M. J., Chellingsworth, M. C., Jubb, R., Thawley, A. R., Undre, N. A., Kill, D. C.: A pharmacokinetic study of the active metabolite of nabumetone in young healthy subjects and older arthritis patients. Eur J Clin Pharmacol. 1989; 36, 299–305.
7. Davies, N. M.: Clinical pharmacokinetics of nabumetone. The dawn of selective cyclo-oxygenase-2 inhibition? Clin Pharmacokinet. 1997; 33, 404–416.
8. Patel, B. N., Sharma, N., Sanyal, M., Prasad, A., Shrivastav, P. S.: High-throughput LC-MS/MS assay for 6-methoxy-2-naphthylacetic acid, an active metabolite of nabumetone in human plasma and its application to bioequivalence study. Biomed Chromatogr. 2008; 22, 1213–1224.
9. Srinivas N. R.: Applicability of LC/MS/MS assay for 6-methoxy-2-napthylacetic acid to support the bioequivalence study of nabumetone-comments on the research work of Patel et al. (2008). Biomed Chromatogr. 2009; 23, 674–675.
10. Food and Drug Administration: FDA Guidance for Industry: Bioanalytical Method Validation. 2001; http://www.fda.gov/cder/quidance (15. 11. 2010).
Štítky
Farmácia FarmakológiaČlánok vyšiel v časopise
Česká a slovenská farmacie

2011 Číslo 1
Najčítanejšie v tomto čísle
- Životní jubileum doc. PharmDr. Miloslavy Rabiškové, CSc.
- K výuce chemie farmaceutických pomocných látek v rámci studia farmacie v České republice a ve Slovenské republice
- Studium ledvinné akumulace 99mTc-MAG3 a 99mTc-DMSA s využitím in vitro buněčného modelu
- Pracovní den sekce technologie lékůČeské farmaceutické společnosti ČLS JEPPokroky v lékových formách