
Absence of Intestinal PPARγ Aggravates Acute Infectious Colitis in Mice through a Lipocalin-2–Dependent Pathway
To be able to colonize its host, invading Salmonella enterica serovar Typhimurium must disrupt and severely affect host-microbiome homeostasis. Here we report that S. Typhimurium induces acute infectious colitis by inhibiting peroxisome proliferator-activated receptor gamma (PPARγ) expression in intestinal epithelial cells. Interestingly, this PPARγ down-regulation by S. Typhimurium is independent of TLR-4 signaling but triggers a marked elevation of host innate immune response genes, including that encoding the antimicrobial peptide lipocalin-2 (Lcn2). Accumulation of Lcn2 stabilizes the metalloproteinase MMP-9 via extracellular binding, which further aggravates the colitis. Remarkably, when exposed to S. Typhimurium, Lcn2-null mice exhibited a drastic reduction of the colitis and remained protected even at later stages of infection. Our data suggest a mechanism in which S. Typhimurium hijacks the control of host immune response genes such as those encoding PPARγ and Lcn2 to acquire residence in a host, which by evolution has established a symbiotic relation with its microbiome community to prevent pathogen invasion.
Published in the journal:
Absence of Intestinal PPARγ Aggravates Acute Infectious Colitis in Mice through a Lipocalin-2–Dependent Pathway. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003887
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003887
Summary
To be able to colonize its host, invading Salmonella enterica serovar Typhimurium must disrupt and severely affect host-microbiome homeostasis. Here we report that S. Typhimurium induces acute infectious colitis by inhibiting peroxisome proliferator-activated receptor gamma (PPARγ) expression in intestinal epithelial cells. Interestingly, this PPARγ down-regulation by S. Typhimurium is independent of TLR-4 signaling but triggers a marked elevation of host innate immune response genes, including that encoding the antimicrobial peptide lipocalin-2 (Lcn2). Accumulation of Lcn2 stabilizes the metalloproteinase MMP-9 via extracellular binding, which further aggravates the colitis. Remarkably, when exposed to S. Typhimurium, Lcn2-null mice exhibited a drastic reduction of the colitis and remained protected even at later stages of infection. Our data suggest a mechanism in which S. Typhimurium hijacks the control of host immune response genes such as those encoding PPARγ and Lcn2 to acquire residence in a host, which by evolution has established a symbiotic relation with its microbiome community to prevent pathogen invasion.
Introduction
Salmonella enterica serovar Typhimurium is a Gram-negative, facultative intracellular pathogen that causes a wide array of disorders ranging from systemic disease to enterocolitis in multiple hosts [1]. In mice, S. Typhimurium normally causes a disease that resembles systemic typhoid fever. However, compromising the gut microbiome with antibiotics prior to S. Typhimurium infection in mice has been used to mimic salmonellosis in humans, which involves increased S. Typhimurium colonization of the intestine coupled with a marked host-induced inflammatory response leading to colitis [2]. Recent reports indicate that this massive inflammatory response elicited by S. Typhimurium is associated with increased secretion of the interleukins IL-17 and IL-22 [3], [4], which are critical components of mucosal immunity to bacterial pathogens in the gut. In particular, the IL-17/IL-22 axis mediates the recruitment of antimicrobial peptides from the intestinal epithelial compartment, including lipocalin-2 (Lcn2) [3]–[7]; these peptides dramatically affect the gut microbiota.
Lcn2 (also known as SIP24, 24p3, NGAL, uterocalin, and siderocalin) was first co-purified and found to be covalently associated with human neutrophil gelatinase (matrix metalloproteinase (MMP)-9) [8], [9]. This association between Lcn2 and MMP-9 has been shown to protect MMP-9 from degradation and to preserve its enzymatic activity [8], [10]. In addition, Lcn2 functions in mammalian innate immunity by chelating bacterial siderophores, thereby sequestering iron from bacteria and inhibiting their growth [11], [12]. Intriguingly, S. Typhimurium appears to be resistant to Lcn2, since its population in the intestinal milieu expands dramatically during inflammation [4], [13], [14]. This unique strategy is achieved by genes such as those in the iroN iroBCDE gene cluster, which encodes salmochalin, a siderophore that does not bind Lcn2 [4], [13], [15], [16], thus conferring a competitive advantage to S. Typhimurium over other microbes during growth in the inflamed gut.
Recently, peroxisome proliferator-activated receptor gamma (PPARγ) has been shown to be regulated by a number of bacterial pathogens including Helicobacter pylori and Mycobacterium tuberculosis [17]–[19], greatly impacting disease severity. PPARγ is a member of the nuclear receptor superfamily of ligand-dependent transcription factors and is predominantly expressed in adipose tissue and colonic epithelium [20], [21]. Expression has also been detected in colonic macrophages and in T and B cells of humans and rodents [20], [22]. PPARγ has been proclaimed to be a master regulator of inflammation, a role that is achieved in part by antagonizing the activities of the transcription factors AP-1, STAT, and NFκB [23], [24]. In vivo studies have demonstrated that PPARγ ligands actively suppress the inflammatory response by attenuating the production of chemokines and cytokines secreted from epithelial cells, macrophages, and T and B lymphocytes [23]–[26].
The role of PPARγ in the etiology and treatment of colitis has been of great interest, because its ligands have long been used to treat type-2 diabetes and are known to decrease the severity of colitis induced in mouse models [27]–[33]. Moreover, PPARγ+/− heterozygous mice exhibit increased susceptibility to experimentally induced colitis, indicating PPARγ's involvement in maintaining gut homeostasis [28], [33]. Furthermore, the observation that intestinal epithelium-specific ablation of PPARγ aggravates dextran sodium sulfate (DSS)-induced colitis demonstrates the strong influence of intestine-derived PPARγ on colitis severity [20]. Studies in human subjects have revealed that colonic epithelial cells from ulcerative colitis patients display drastically reduced expression of PPARγ, suggesting that its presence in gut epithelium may have a protective effect against colonic inflammation in humans [29]. Despite these observations, the role of PPARγ in S. Typhimurium-induced infectious colitis remains unknown.
In this study, we explored whether S. Typhimurium regulates host PPARγ levels during infectious colitis and evaluated PPARγ's contributions to the etiology of the disease. Our data reveal that S. Typhimurium inhibits PPARγ expression in the intestinal epithelium, which triggers a massive innate immune response that includes expression of Lcn2. Selective epithelial ablation of PPARγ dramatically increased Lcn2 expression and its secretion after S. Typhimurium challenge, confirming the importance of epithelium-derived PPARγ in colitis. Furthermore, increased secretion of Lcn2 stabilized MMP-9 via direct protein-protein interaction, which further aggravated the colitis. Finally, we demonstrate that Lcn2-null mice exposed to S. Typhimurium displayed significantly less-severe colitis.
Results
Regulation of PPARγ in the mouse colon by S. Typhimurium
Although PPARγ signaling controls various cellular processes during inflammation and pathogenesis, its regulation during S. Typhimurium-induced colitis remains unexplored. To gain insight into PPARγ's role in infectious colitis, streptomycin-pretreated C57BL/6 mice were infected with S. Typhimurium. An incubation period of 24 h was deliberately selected to evaluate the early phase of PPARγ response, which is crucial for further downstream effector regulation. Strikingly, S. Typhimurium infection resulted in ∼60% down-regulation of PPARγ gene expression in the colon, as detected by real-time PCR (Fig. 1A). Immunoblotting revealed a similar down-regulation of PPARγ expression at the protein level (Fig. 1B) in the colon 24 h after infection. As expected, this down-regulation resulted in a marked reduction in the DNA-binding activity of PPARγ in colonic cells in vivo (Fig. 1C), thus reducing PPARγ's tight control over its potential targets. A similar effect was detected in the cecum of infected mice (Fig. S1A–C).
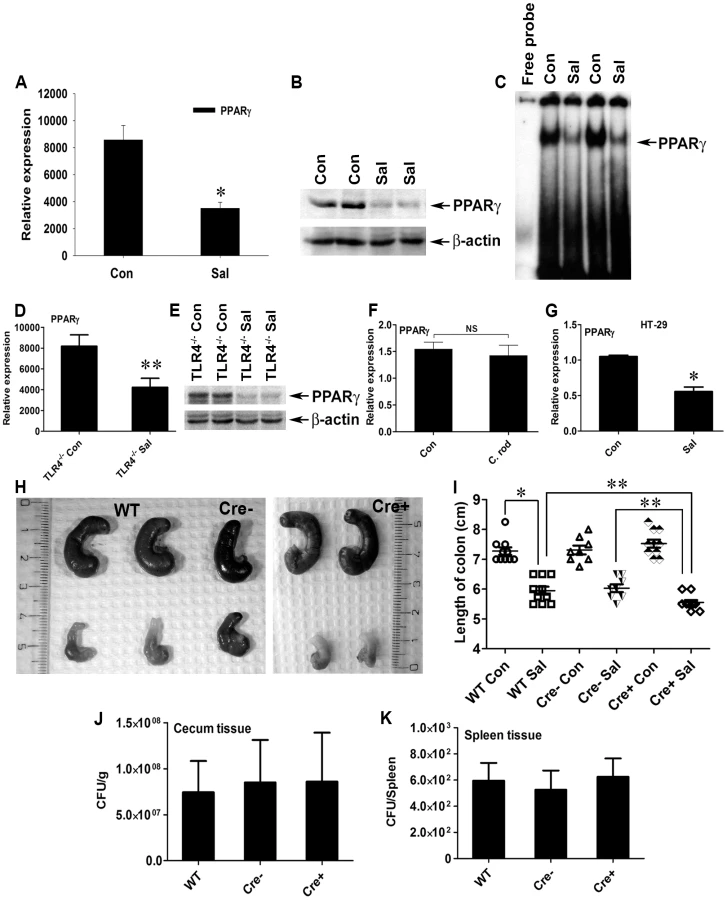
Since a previous study reported Toll-like receptor (TLR)-4-dependent PPARγ regulation by microbial lipopolysaccharide in macrophages [34], we asked whether this negative regulation of PPARγ by S. Typhimurium was TLR-dependent. A significant increase in the expression of gene encoding TLR-4, but not TLR-2 and TLR-5, was observed in the infected mice, suggesting that TLR4 may be involved in regulating PPARγ (Fig. S2A–C). However, S. Typhimurium infection in TLR-4−/− mice resulted in a similar decline in PPARγ expression (Fig. 1D and E), suggesting that this PPARγ regulation was independent of TLR-4. However, the expression of TLR-2 and TLR-5 gene in the infected TLR-4−/− mice remained unchanged, negating the possibility of their increased activity in the absence of TLR-4 (Fig. S2D and E). To confirm the specificity of PPARγ deregulation by S. Typhimurium, another potential gut pathogen, Citrobacter rodentium, was used to infect mice. C. rodentium was unable to alter host PPARγ levels, confirming that the process of PPARγ down-regulation by S. Typhimurium was not a general effect (Fig. 1F).
Next, we set out to identify the cell types that predominantly respond to S. Typhimurium. A sharp decrease in the gene expression of PPARγ occurred when cultures of colonic epithelial cells (HT-29 cells) were infected with S. Typhimurium, suggesting the importance of the intestinal epithelium in this process (Fig. 1G). These observations not only indicate that S. Typhimurium infection directly impacts PPARγ levels in the colonic epithelium, but also suggest that PPARγ's pivotal role in homeostasis within the intestinal tract affects infectious colitis.
Specific ablation of PPARγ in the intestinal epithelium aggravates S. Typhimurium-induced colitis
To better understand the implications of this PPARγ regulation by S. Typhimurium, we bred mice harboring a floxed Pparγ (PPARγfl/fl) to mice expressing the Cre transgene under control of the promoter of the villin gene. These mice, in which cre recombinase mediated the targeted disruption of PPARγ in intestinal epithelial cells, were designated PPARγVillinCre+ mice and were used in parallel with littermate control PPARγVillinCre - or wild-type (C57BL/6) mice. Interestingly, S. Typhimurium infection resulted in more severe colitis in the PPARγVillinCre+ mice compared to PPARγVillinCre - or wild-type mice at 24 h (Fig. 1H and I). Shortening and thickening of the cecum and the colon, which are hallmarks of colitis, were much more pronounced in PPARγVillinCre+ mice than in wild-type or PPARγVillinCre - mice (Fig. 1I). Of note, the S. Typhimurium-infected TLR-4−/− mice and the wild-type (C57BL/6) mice infected with C. rodentium exhibited significantly shortened colons, indicating active colitis (Fig. S2F and G).
S. Typhimurium cells were recovered in similar numbers from cecum tissue and from the spleens of wild-type, PPARγVillinCre-, and PPARγVillinCre+ mice 24 h after infection (Fig. 1J and K), indicating that infection in the cecum as well as systemic dissemination of S. Typhimurium were comparable between these groups. Histological analysis further revealed that apart from increased infiltration of inflammatory cells, tissue damage was more in the colons of PPARγVillinCre+ mice compared to PPARγVillinCre - mice and to wild-type mice after infection (Fig. 2). Notably, similar levels of the epithelial cell markers villin 1, cytokeratin 8, and cytokeratin 20 were detected in infected and mock-infected colonic samples, confirming that the ratios of epithelial cells in the colonic extracts from these groups were consistent and did not account for the reduced levels of PPARγ (Fig. S3). These results were consistent with previous reports indicating a protective role of PPARγ in the intestinal epithelium in experimental inflammatory bowel disease [20]. However, the molecular mechanisms underlying these observations remain elusive.

Depletion of epithelial PPARγ in the colon triggers an elevated immune response during infectious colitis
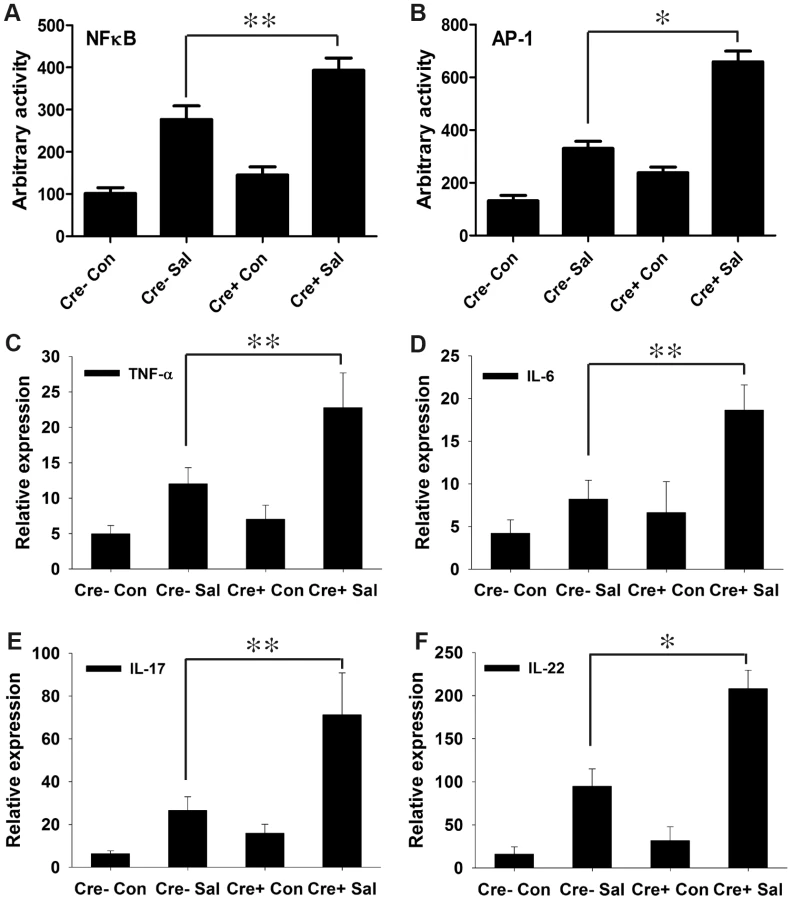
We previously demonstrated the direct participation of PPARγ in host-microbe crosstalk and the consequent regulation of innate immune functions [35], [36]. Moreover, PPARγ has been proposed to regulate inflammation by antagonizing the NFκB and AP-1 pathways [23], [24], which in turn may modulate immune responses. To test this hypothesis in infectious colitis, we analyzed the activities of NFκB and AP-1 via electromobility shift assay using nuclear extracts from colonic scrapings. As anticipated, depletion of epithelial PPARγ was coupled with a marked increase in the activities of NFκB and AP-1 in the colon after S. Typhimurium infection compared to littermate control (PPARγVillinCre-) mice (Fig. 3A and B, Fig. S4).
A potential challenge was to obtain maximal numbers of epithelial cells in colonic scrapings; epithelial cells contribute the majority of colonic PPARγ, but macrophages and B and T lymphocytes also produce it [20], [22]. To overcome this problem, we used mucosal scrapings from the colon and tested for the presence of PPARγ transcripts through real-time PCR. Minimal expression and activity of PPARγ was detected in mock-infected and infected PPARγVillinCre+ mice compared to littermate control PPARγVillinCre - and wild-type (C57BL/6) mice (Fig. S5A and B). This finding was further validated in cecum scrapings (Fig. S1D). Thus, it was evident that epithelial cells were the major cell type in our samples; the presence of infiltrating macrophages and T and B cells in the colonic scrapings was possible, but minimally contributed to PPARγ production. Furthermore, no significant differences in the expression or activity of PPARγ were detected between PPARγVillinCre - and wild-type C57BL/6 mice (Fig. S5), justifying the use of PPARγVillinCre - mice as controls in subsequent experiments.
We next investigated the effect of increased NFκB and AP-1 activities on key regulators of inflammation. After infection, the expression levels of TNF-α and IL-6 in the colon were two-fold higher in PPARγVillinCre+ mice compared to PPARγVillinCre - mice (Fig. 3C and D). IL-6 is a key regulator of the innate T helper type 17 (TH17) response, a critical component of mucosal immunity to intestinal pathogens [3]. Consistent with this role, we observed substantial increases in the expression of IL-17 and IL-22, a typical innate TH17 response signature, in infected PPARγVillinCre+ mice compared to infected PPARγVillinCre - mice, as assessed via real-time PCR (Fig. 3E and F). IL-17 and IL-22 have been linked with intestinal innate epithelial defense mechanisms through the production of antimicrobial peptides [3], [5]–[7]. The expression of Lcn2, a principal target of IL-17 and IL-22 [3], [4], increased by approximately 7-fold in the colons of S. Typhimurium-infected PPARγVillinCre - mice compared to mock-infected mice, an increase that rose to a striking ∼21-fold change in infected PPARγVillinCre+ mice (Fig. 4A). This increase in Lcn2 expression in infected colons was also noticeable at the protein level (Fig. 4B and C), further confirming a hyper-magnified Lcn2 response in infected PPARγVillinCre+ mice. Lcn2 was similarly elevated in the cecum of these mice (Fig. S1E). Interestingly, TLR-4−/− mice infected with S. Typhimurium also showed a significant increase in Lcn2 expression in the colon compared to mock-infected mice (Fig. S2H).

The expression of regenerating islet-derived 3 gamma (Reg3γ), another potent member of the host antimicrobial arsenal and a potential target of IL-17 and IL-22 [3], [37], was also tested. The expression of Reg3γ in colon followed a pattern similar to that of Lcn2 in PPARγVillinCre - and PPARγVillinCre+ mice (Fig. 4D), thus establishing a heightened innate immune response to S. Typhimurium in the absence of epithelial PPARγ. Moreover, S. Typhimurium induced a significant increase in the expression and secretion of Lcn2 (Fig. 4E and F) in parallel with PPARγ down-regulation (Fig. 1G) in colonic epithelial cells, further confirming the importance of epithelial cells in this process. To test whether this Lcn2 up-regulation was a cell-autonomous effect of PPARγ in epithelial cells, we next applied PPARγ small interfering RNA (siRNA) to HT-29 cells. A ∼50% reduction in PPARγ levels (Fig. 4G) led to a significant increase in Lcn2 expression and secretion in the absence of S. Typhimurium infection (Fig. 4H and I). Moreover, when these PPARγ-siRNA treated HT-29 cells were infected with S. Typhimurium, the expression profile of Lcn2 resembled to that observed in vivo (Fig. S2I). These observations not only confirmed the existence of a direct link between PPARγ and Lcn2, but also suggested that Lcn2 regulation by PPARγ may occur locally in epithelial cells without the intervention of other cell types.
At this juncture, the observed dichotomy in PPARγ's role in infectious colitis was surprising. Although we initially observed that the absence of PPARγ led to increased colitis severity (Fig. 1H and I; Fig. 2), in sharp contrast we also detected a simultaneous elevated innate immune response that seemed to serve a protective role. Taken together, these results reflect PPARγ's tight control over intestinal homeostasis in the host during S. Typhimurium pathogenesis. We next questioned the rationale behind this heightened Lcn2 expression, given that S. Typhimurium is typically resistant to Lcn2's antimicrobial activity [4], [13], [15], [16], and speculated that Lcn2 may have a more diverse role in the disease process.
Lcn2 promotes MMP-9 stability and contributes to colitis severity
Having established that Lcn2 expression was markedly increased in PPARγVillinCre+ mice (Fig. 4A–C), we next sought to dissect its possible role in the increased colonic damage observed in PPARγVillinCre+ mice in the absence of PPARγ (Fig. 2), suggesting ongoing exaggerated protease action. Interestingly, Lcn2 has been shown to increase the stability of MMP-9 by protecting it from degradation, resulting in an increase in its enzymatic activity independent of transcriptional regulation [8], [10], [38], [39]. To test this hypothesis, we analyzed the activity of gelatin agarose-purified secreted gelatinases by zymography using phosphate-buffered saline (PBS) extracts of colonic scrapings of mock - or S. Typhimurium-infected mice. Interestingly, we detected a ∼140 KDa band representative of Lcn2-bound proMMP-9 (proMMP-9/Lcn2) in S. Typhimurium-infected PPARγVillinCre - mice that peaked to an ∼6-fold increase in infected PPARγVillinCre+ mice (Fig. 5A and B). Surprisingly, this increase in the proMMP-9/Lcn2 band in PPARγVillinCre+ mice was associated with a massive increase in the activity of proMMP-9 (detected by zymography Fig. 5A), indicating an increased stability of proMMP-9. The activity of proMMP-9 increased by ∼6 fold in PPARγVillinCre - mice and ∼15 fold in PPARγVillinCre+ mice after infection, while MMP-9 activity rose from ∼4 fold in PPARγVillinCre - mice to ∼7 fold in PPARγVillinCre+ mice after infection (Fig. 5A and B). In contrast, MMP-2 activity remained almost unchanged between groups. Importantly, no significant difference in MMP-9 gene expression occurred between the PPARγVillinCre - and PPARγVillinCre+ infected groups, as detected by real-time PCR (Fig. 5C), which confirmed that the differences in MMP-9 activity were independent of transcriptional regulation. Furthermore, MMP-2 expression was similar between the infected and mock-infected groups (Fig. 5D). No noticeable difference in the expression of TIMP-1, the endogenous inhibitor of MMP-9, was observed between the infected mice groups (Fig. 5E), eliminating the possibility of its involvement in the observed deregulation of MMP-9 activity.

To further confirm this phenomenon, we next analyzed the protein levels of MMP-9 and Lcn2 by immunoblotting gelatin-agarose-purified PBS extracts of colonic scrapings under non-reducing conditions. As anticipated, the ∼140 KDa proMMP-9/Lcn2 band, the level of which increased by ∼5 fold in infected PPARγVillinCre - mice, peaked at ∼15-fold in PPARγVillinCre+ infected mice (Fig. 5F and G). This increase led to an ∼15-fold increase in proMMP-9 protein levels in PPARγVillinCre - infected mice and reached a striking increase of ∼40 fold in PPARγVillinCre+ infected mice, while MMP-9 climbed ∼5 fold in PPARγVillinCre+ infected mice (Fig. 5F and G). When the gels were re-probed for Lcn2, bands at exactly the same location as the ∼140 KDa band, characteristic of proMMP-9 bound to Lcn2, were detected (Fig. 5F and G). These results indicate that Lcn2 is secreted from the intestinal epithelium, in accordance with previous reports [3], and more importantly confirm the binding of Lcn2 to secreted proMMP-9 in the intestinal milieu, which is pivotal for subsequent pathological processes. However, since the increase in the levels of the Lcn2/MMP-9 complex in the intestinal milieu correlates with the increased infiltration of neutrophils it is possible that these cells may contribute to the observed elevation of Lcn2/MMP-9. It is noteworthy that the ∼6-fold increase in Lcn2 secretion in S. Typhimurium-infected PPARγVillinCre+ mice (Fig. 5F and G) did not precisely correspond to its ∼15-fold increase at the expression level (Fig. 4A–C), because it represented only the Lcn2 fraction bound to proMMP-9 during gelatin-agarose purification of secreted gelatinases. Reasonably, no low molecular-weight band for Lcn2 alone was detected (Fig. 5F). It may also be noted that the proportion of bound or free Lcn2 or proMMP-9 at any given point would largely depend on the availability, stability, and importantly the stoichiometry of binding between these molecules. Taken together, these results conclusively indicate that lack of epithelial PPARγ substantially elevates Lcn2 expression and its secretion in the intestinal milieu during S. Typhimurium infection, resulting in increased MMP-9 stabilization and activity.
Absence of Lcn2 significantly protects mice from S. Typhimurium-induced colitis
To validate the observed involvement of Lcn2 in S. Typhimurium-induced colitis, we next checked for colitis induction in streptomycin-pretreated Lcn2−/− mice mock - or S. Typhimurium-infected for 24 h. As expected from the above observations, marked reductions in the extent and severity of S. Typhimurium-induced colitis were observed in mice devoid of Lcn2 (Fig. 6A and B). Shortening and thickening of the cecum and colon were considerably restricted in the Lcn2−/− mice 24 h after S. Typhimurium challenge (Fig. 6A and B). These results were confirmed by observations of reduced thickening of the mucosa and sub-mucosa, with negligible tissue damage (Fig. 6C–H). However, a modest increase in infiltrating cells and consequent myeloperoxidase (MPO) activity was noted (Fig. 6G and H). Moreover, the number of S. Typhimurium in the cecum and spleen of these Lcn2−/− mice was comparable to that of wild-type mice after 24 h (Fig. 6I and J). As expected, Lcn2 expression was not detected in these mice by real-time PCR (Fig. S6A) or by immunoblotting (Fig. S6B) of the colons of Lcn2−/− mice. Thus, these observations highlight Lcn2's unique role in the induction and severity of infectious colitis.

To more precisely track the molecular mechanisms active in Lcn2−/− mice after S. Typhimurium infection, we assessed the secretion of gelatinases in the colon. We observed an ∼3.5-fold increase in proMMP-9 activity in Lcn2−/− mice after infection versus mock infection (Fig. 7A and B), compared to a ∼6-fold increase in PPARγVillinCre - infected mice (Fig. 5A and B). No notable differences in the activities of MMP-9 and MMP-2 were detected between groups. The basal secretion of gelatinases was similar in PPARγVillinCre - and Lcn2−/− mock-infected mice (Fig. S7A), justifying the comparison between these groups in this case.

We next examined the levels of secreted MMP-9 using gelatin-purified colonic extracts of mock - or S. Typhimurium-infected Lcn2−/− mice. Compared to the ∼15-fold increase in secreted proMMP-9 that was observed in PPARγVillinCre - mice after infection (Fig. 5F and G), a mere ∼3-fold increase in the levels of secreted proMMP-9 was detected in Lcn2−/− mice after infection, suggesting reduced extracellular stability and possible degradation of MMP-9 protein in the absence of Lcn2 (Fig. 7C and D). No significant difference in the colonic expression of MMP-9 was detected between Lcn2−/− and Lcn2+/+ mice after infection (Fig. 7E), confirming that the differences in the protein levels of MMP-9 were due to impaired stability. Moreover, colonic expression of MMP-2 and TIMP-1 was similar in Lcn2+/+ and Lcn2−/− mice after infection (Fig. S7B and C), excluding their involvement in this process.
To investigate any possible mechanistic differences between the Lcn2−/− and Lcn2+/+ mice during S. Typhimurium infection, we validated several key regulators involved in the pathophysiology of infectious colitis. Real-time PCR revealed no appreciable differences in the down-regulation of PPARγ by S. Typhimurium between Lcn2−/− and Lcn2+/+ mice (Fig. S8A), confirming that PPARγ regulation by S. Typhimurium was essentially the same in Lcn2−/− mice. Consequently, the expression levels of TNF-α and IL-6 were similar between the infected groups (Fig. S8B and C). Colonic expression of IL-17 and IL-22 also exhibited negligible differences between Lcn2−/− and Lcn2+/+ infected mice (Fig. S8D and E), as did Reg3γ expression (Fig. S8F). Together, these observations suggest that the mechanistic chain of events during S. Typhimurium infection in wild-type and Lcn2−/− mice was fundamentally similar; the absence of Lcn2 exclusively conferred protection to these mice against S. Typhimurium-induced colitis.
To determine whether the regulation of PPARγby S. Typhimurium is a transient effect or whether the Lcn2−/− mice are protected at later stages of infection, we infected PPARγVillinCre-, PPARγVillinCre+, Lcn2−/−, and Lcn2+/+ mice with S. Typhimurium and sacrificed them after 72 h. Long-term S. Typhimurium infection resulted in more severe colitis in PPARγVillinCre+ mice compared to PPARγVillinCre - mice, while Lcn2−/− mice were significantly protected against colitis (Fig. S9A); colonic shortening displayed a similar profile (Fig. S9B). There was no significant difference in the number of S. Typhimurium recovered from the cecum or spleen of these mice 72 h after infection (Fig. S9C and D). Histological analysis revealed more severe colitis in PPARγVillinCre+ mice than in PPARγVillinCre - mice, with significantly increased neutrophil infiltration and MPO activity (Fig. S9E-L and S9U-W). Although the Lcn2−/− mice exhibited moderate colitis, they were associated with marked reductions in neutrophil infiltration, edema, and MPO activity compared to the rest of the groups 72 h after infection (Fig. S9M–W). Interestingly, PPARγ expression was still significantly reduced after 72 h in the colons of PPARγVillinCre - mice, confirming that PPARγ regulation by S. Typhimurium was not a transient event (Fig. S9X and Y).
Discussion
Intestinal pathogens employ diverse strategies to modulate the host environment in order to survive in this competitive niche. The approaches that individual pathogens adopt depend largely on the tenure of their residence in the host. For instance, H. pylori up-regulates host PPARγ as part of a feedback mechanism to suppress exaggerated inflammation, ensuring its unperturbed long-term survival in the host [17], [19]. Similarly, M. tuberculosis induces PPARγ expression in infected individuals and subsequently interacts with host PPARγ by modulating macrophage function for its survival [18]. Here, we unravel a novel mechanism used by S. Typhimurium to down-regulate PPARγ in the intestinal epithelium, initiating acute inflammation via the host immune and protease machinery, thereby transforming the intestine into a more hostile niche where it is best adapted to survive and outgrow its competitors. This TLR-4-independent regulation of PPARγ by S. Typhimurium seems to be characteristic of this pathogen, since C. rodentium infection, did not alter PPARγ levels.
PPARγ activation has been shown to ameliorate the severity of inflammatory bowel disease in rodent DSS, trinitrobenzene sulphonic acid, and ischemic colitis models [20], [27], [30], [32], [33]. Since PPARγ is expressed in epithelial cells as well as in immune cells infiltrating colonic tissue during inflammation, the cell type that mainly contributes to PPARγ production during colitis remains a point of contention. Interestingly, clinical reports indicate that PPARγ expression in colonic epithelium is impaired in ulcerative colitis patients, while its expression in inflammatory cells remains normal [40]. This observation was corroborated by Adachi et al., who reported that PPARγ expressed in the colonic epithelium has an endogenous role in protection against DSS-induced colitis [20]. These findings, together with our data in Fig. 1G, demonstrating PPARγ regulation by S. Typhimurium in human colonic epithelial cells, prompted us to use epithelial-specific PPARγ-null mice to unravel the chain of events that occur during S. Typhimurium-induced colitis. This strategy allowed us to magnify the subtle molecular changes induced by S. Typhimurium in the host via down-regulation of epithelial PPARγ. S. Typhimurium induced much more severe colitis in these mice (Fig. 1H and I; Fig. 2), highlighting, for the first time, the importance of intestinal epithelium-derived PPARγ in protection against bacterial pathogenesis.
Our data demonstrate that S. Typhimurium-induced depletion of epithelial PPARγ uncouples PPARγ's tight control over the inflammatory transcription factors NFκB and AP-1, resulting in the release of the pro-inflammatory cytokines TNF-α and IL-6. This influx of pro-inflammatory cytokines from the intestinal epithelium initiates an acute-phase immune response characterized by elevated expression of IL-17 and IL-22. These results are consistent with a recent report by Geddes et al. of the induction of the innate TH17 response by S. Typhimurium during the early phases of infection [3]. Collectively, our results imply that these inflammatory signaling circuits are orchestrated by S. Typhimurium during the early phases of infection via the regulation of epithelial PPARγ, which is pivotal for the entire process.
The secretion of IL-17 and IL-22 in the inflamed colon, which is initiated by S. Typhimurium, has been shown to facilitate the production of antimicrobials, including Lcn2, from the intestinal epithelium [3]–[7]. Our observations confirm the contribution of IL-17 and IL-22 to Lcn2 production and secretion, but also suggest that S. Typhimurium-induced increases in NFκB and AP-1 activity in epithelial cells via PPARγ down-regulation may directly influence Lcn2 expression in the same cells independent of IL-17 and IL-22 (Fig. 7F). This hypothesis was further corroborated by the induction of Lcn2 in non-infected human colonic epithelial cells treated with siRNA directed against PPARγ. It seems reasonable to assume that S. Typhimurium may utilize this more direct pathway of Lcn2 regulation, since Lcn2 expression is known to be regulated by NFκB or even AP-1 [41], [42]; importantly, this entire sequence of events may occur in affected epithelial cells.
Understanding the rationale behind the substantial increase in Lcn2 secretion from the intestinal epithelium during S. Typhimurium infection, which typically utilizes salmochalin, a siderophore resistant to Lcn2 action [4], [13], [15], [16], for iron uptake, was perhaps the biggest challenge in this study. Here, we documented that S. Typhimurium-induced elevated influx of secreted Lcn2 in the intestinal milieu leads to stabilization and a significant increase in MMP-9 activity, through direct extracellular protein-protein binding (Fig. 7F). MMP-9, a member of a family of zinc-dependent endopeptidases that have broad substrate specificity, has been shown to play a pivotal role in the degradation and remodeling of the extracellular matrix during bacterial pathogenesis [19], [43]. Moreover, MMP-9-null mice exposed to DSS or to S. Typhimurium were previously significantly protected from colitis [44], [45], confirming MMP-9's importance in the etiology of the disease. Although MMPs are secreted by a variety of cell types, such as fibroblasts, epithelial cells, endothelial cells, neutrophils, macrophages, and lymphocytes, MMP-9 is predominantly expressed in epithelial cells and in inflammatory cells during colitis [44], [46]. Our data suggest that this crosstalk between Lcn2 and MMP-9 in the inflamed gut, which results in increased MMP-9 activity, was crucial for the deleterious impact on the intestinal mucosa observed during infectious colitis. This novel mechanism by which S. Typhimurium exploits the host Lcn2 and MMP-9 synergy to aggravate inflammation and colitis severity is critical, as it calls for the reinterpretation of studies on microbial pathogenesis; other potential pathogens may also employ similar mechanisms.
Interestingly, mice lacking Lcn2 were considerably protected against S. Typhimurium-induced colitis even at the later stages of infection, confirming the key role of this secreted protein in S. Typhimurium pathogenesis. We detected no significant differences in the overall mechanism acting in Lcn2-null and wild-type infected mice, with the exception of a decrease in MMP-9 stability and activity in the colon. Our observations of Lcn2-null mice exposed to S. Typhimurium, which indicated increased expression of IL-17, IL22, and TNF-α, are consistent with the findings of Raffatellu et al. [4]. We also noted moderate inflammation in the colon during the later stages of infection. However, disease severity may be impacted by differences in experimental setup, including S. Typhimurium strains, bacterial load and phase of growth in the inoculum, and importantly, differences in intestinal microflora between mice.
In conclusion, our investigation unveiled a novel pathogenic mechanism utilized by S. Typhimurium to thrive and to induce colitis in its host. This study motivates the development of therapeutic interventions directed against this Lcn2-dependent, MMP-9-driven tissue degradation pathway to combat salmonellosis. However, research aimed toward a better understanding of the pathogenic mechanisms of S. Typhimurium or other pathogens in the gut remains an exciting area for future studies.
Materials and Methods
Ethics statement
All protocols involving animals were approved by the Regional Animal Research Ethical Board, Stockholm, Sweden, following proceedings described in European Union legislation (Council Directive 86/609/EEC). Animal husbandry was in accordance with institutional guidelines at Karolinska Institutet and was approved by the above-mentioned ethical board (Stockholms norra djurförsöksetiska nämnd, Ref: N 100/10).
Bacterial strains, culture and colonization assessment
The naturally streptomycin-resistant wild type strain S. enterica serovar Typhimurium SL1344 [47], a generous gift from Prof. Mikael Rhen was used in this study. Naturally occurring naldixic acid-resistant Citrobacter rodentium, DBS100 (ATCC 51459) was also used for mouse infection. Prior to inoculation into host mice, strain SL1344 and C. rodentium were grown overnight at 37°C in Luria-Bertani (LB) broth, diluted 1∶20 in fresh medium, and sub-cultured for 3–4 h under mild aeration until an optical density of 0.4 to 0.6 at 600 nm was reached. Bacteria were washed twice in cold PBS and then suspended in cold PBS for mouse inoculation.
The cecum and spleen from post-sacrifice mice were collected in 1 mL of sterile PBS. Samples were kept on ice, minced, and homogenized. Serial dilutions of the homogenates were plated on LB agar plates supplemented with 100 µg/mL streptomycin to enumerate S. Typhimurium. Plates were incubated overnight at 37°C, and colonies were counted thereafter.
Cell-culture assays
The human epithelial cell line HT-29 (ATCC-HTB-38) was obtained from the American Type Culture Collection. Cells were grown in RPMI 1640 (Invitrogen) medium supplemented with 10% heat-inactivated fetal calf serum (Invitrogen). Cells were maintained in a 37°C humidified atmosphere with 5% CO2. Epithelial morphogenesis was monitored via microscopy; cell densities for each experiment did not exceed 80% to prevent contact inhibition.
For co-culture experiments, cells were treated with S. Typhimurium (0.25×107 cells/well; 10∶1 bacterial cells:eukaryotic cells) for 6 h. Controls were treated with culture medium only. After 6 h medium was removed, cells were washed, fresh medium with 1% penicillin/streptomycin was added, and the cells were incubated for 18 h, after which the cells were collected and lysed.
For siRNA experiments HT-29 cells were plated at a density of 0.0625×105 cells/cm2. Down-regulation of PPARγ transcripts was achieved with SMART Pool siRNA directed against PPARγ (Thermo Scientific). Controls were transfected with non-targeting siRNA (Thermo Scientific) at a final siRNA concentration of 40 nM. Transfection was carried out according to the manufacturer's protocol using DharmaFECT 4 (Thermo Scientific) reagent at a final concentration of 0.3%.
Bacterial infection in mice
Specific pathogen-free C57BL/6 wild-type mice carrying a targeted disruption of the gene encoding PPARγ in intestinal epithelial cells were generated by breeding animals harboring a floxed Pparγ (PPARγfl/fl) [48] to mice expressing the Cre transgene under control of the villin promoter; these mice were designated as PPARγVillinCre+, and their littermate control mice were designated as PPARγVillinCre−. Lcn2-deficient (Lcn2−/−) mice [11], [38] and TLR4−/− mice (Jackson Laboratory Stock No: 007227) aged 8–10 weeks were also used in this study. The Lcn2−/− mice were generated previously [11] and were backcrossed into the C57BL/6 background for at least 10 generations [38]. All experiments were performed under standard controlled conditions and all efforts were made to minimize animal suffering.
Groups of mice were pretreated with streptomycin (0.1 mL of a 200 mg/mL solution in sterile water) orally 24 h prior to either mock (PBS) or S. Typhimurium (1×108 colony-forming units/mouse) inoculation via gavage. At 24 h or 72 h after infection, mice were euthanized and the cecum, spleen, and colon were collected for analysis. For C. rodentium infection, mice were given metronidazole at 750 mg/L for 4 days, which was withdrawn prior to inoculation with 1×109 colony-forming units/mouse via gavage. C. rodentium-infected mice were sacrificed 6 days after infection [49]. Colon lengths were measured using a centimeter scale.
Tissue extraction, partial purification of gelatinases, and zymography
The mucosal layer of the colon was carefully scraped and suspended in PBS containing protease inhibitors (Roche), minced, and centrifuged at 6000 g for 15 min. The supernatant was collected for use as PBS extracts for the analysis of secreted proteins, while the pellet was re-extracted in lysis buffer (10 mM Tris-HCl [pH 8], 150 mM NaCl, 1% Triton X-100, and protease inhibitors) to obtain Triton X-100 extracts [41]. For partial purification of MMP-9 and MMP-2, PBS extracts of the respective samples were incubated with gelatin-agarose beads (Sigma) at 4°C for 1 h followed by centrifugation at 1500 rpm. The supernatant was collected as flow-through and used as a loading control. The pellet was washed twice with PBS through centrifugation at 1500 rpm and the gelatinases were eluted in Lammeli sample loading buffer. For assays of MMP-9 and MMP-2 activity, gelatin zymography was performed as described previously [43]. Zymographic bands were quantified using LabImage software (KAPELAN).
Immunoblotting
Triton X-100 extracts (50 µg/lane) from colon samples were immunoblotted. Gelatin-agarose-purified PBS extracts were immunoblotted under non-reducing conditions. Cell-culture supernatants were concentrated using a vacuum centrifuge and volumetrically analyzed by immunoblotting.
Immunoblots were probed with anti-PPARγ (Cell Signaling), anti-MMP-9 (Abcam), anti-Lcn2 (Abcam), and anti-β-actin (Santa Cruz Biotechnology) antibodies. Immunodetection with an appropriate secondary peroxidase-conjugated antibody (DAKO) was followed by electrochemiluminescence (Santa Cruz Biotechnology). Quantification of protein bands was performed with the LabImage software. Fold changes were calculated using densitometry values for bands representing proteins of interest, normalized to densitometry values for β-actin bands of respective samples. Representative blots from at least two independent experiments are shown.
Real-time PCR
Total RNA was extracted with an RNeasy Mini Kit (Qiagen), and cDNA was synthesized with SuperScript II (Invitrogen), both procedures according to the manufacturers' protocols. We measured gene expressions with SYBR Green (Applied Biosystems)-based quantitative reverse transcription PCR. Primers were designed and tested according to Applied Biosystems recommendations (Table S1). Sample setups always included at least five biological replicates and experimental triplicates. The changes in mRNA expression of respective samples compared to control were expressed as ΔΔCt = ΔCtcontrol−ΔCtrespective samples (ΔCt = Ct value for the gene of interest - Ct value for β-actin of the respective sample). Relative expressions in the genes in respective samples were calculated as 2ΔΔCt [19].
Electromobility shift assay
Nuclear extracts from colonic scrapings were prepared according the nuclear extraction protocol of Schreiber [50]. DNA binding was assayed with 10 µg of nuclear extract in binding buffer (25 mM HEPES [pH 7.9], 70 mM KCl, 10% glycerol, 5 mM dithiothreitol, and 1 µg polydIdC (Amersham)) in the presence of 50,000 cpm of a radiolabeled oligonucleotide probe. The probe for PPARγ (sequences 5′-TCTCTCTGGGTGAAATGTGC-3′ and 5′-AGAGGCACATTTCACCCAGAGAGA-3′) has high PPARγ-specificity and moderate affinity to ensure weak binding to other PPARs [35]. Probes for NFκB (sequences 5′-GATCCAGAGGGGACTTTCCGAG-3′ and 5′ - TCGACTCGGAAAGTCCCCTCTG-3′) and for AP-1 (sequences 5′-CTGATGACTCAGAG-3′ and 5′-CTCTGAGTCATCAG-3′) were used. polydIdC and probe were added to extracts and incubated for 30 min before gel electrophoresis. Bands were quantified with LabImage.
Histology and pathology scoring
The colons of mock - or S. Typhimurium-infected mice from the respective groups were sectioned for histological studies. Distal part of the colon was used for histological analysis in all cases. Tissue samples were fixed in 10% formalin and embedded in paraffin. Sections (5 µm) were cut with a microtome, stained with hematoxylin and eosin [43], and observed under a Zeiss microscope. Images were captured using Axiovision, LE 64 software (Carl Zeiss Microscopy) at original magnification 5×10 and 20×10 and processed in Adobe Photoshop CS6 (Adobe Systems incorporated). Pathology scoring for neutrophil infiltration and edema was rated from 0 to 5 according to severity, under blinded conditions, by an experienced pathologist (RMB).
MPO activity assay
Whole-cell extracts from colonic scrapings were assayed for MPO activity using the Myeloperoxidase Activity Assay Kit (Invitrogen) following the manufacturer's protocol.
Statistical analysis
Densitometry data were fitted using SigmaPlot 2001 (SPSS) or GraphPad Prism 5 (GraphPad Software). Data are presented as the mean ± standard error of the mean. Between-group comparisons were carried out using either Student's t-test or Student-Newman-Keuls test (ANOVA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BaumlerAJ, TsolisRM, FichtTA, AdamsLG (1998) Evolution of host adaptation in Salmonella enterica. Infection and Immunity 66 : 4579–4587.
2. BarthelM, HapfelmeierS, Quintanilla-MartinezL, KremerM, RohdeM, et al. (2003) Pretreatment of Mice with Streptomycin Provides a Salmonella enterica Serovar Typhimurium Colitis Model That Allows Analysis of Both Pathogen and Host. Infection and Immunity 71 : 2839–2858.
3. GeddesK, RubinoSJ, MagalhaesJG, StreutkerC, Le BourhisL, et al. (2011) Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nature medicine 17 : 837–844.
4. RaffatelluM, GeorgeMD, AkiyamaY, HornsbyMJ, NuccioSP, et al. (2009) Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell host & microbe 5 : 476–486.
5. BettelliE, KornT, OukkaM, KuchrooVK (2008) Induction and effector functions of T(H)17 cells. Nature 453 : 1051–1057.
6. ColonnaM (2009) Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity 31 : 15–23.
7. OuyangW, KollsJK, ZhengY (2008) The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28 : 454–467.
8. KjeldsenL, JohnsenAH, SengelovH, BorregaardN (1993) Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. The Journal of biological chemistry 268 : 10425–10432.
9. TriebelS, BlaserJ, ReinkeH, TschescheH (1992) A 25 kDa alpha 2-microglobulin-related protein is a component of the 125 kDa form of human gelatinase. FEBS letters 314 : 386–388.
10. YanL, BorregaardN, KjeldsenL, MosesMA (2001) The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. The Journal of biological chemistry 276 : 37258–37265.
11. BergerT, TogawaA, DuncanGS, EliaAJ, You-TenA, et al. (2006) Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America 103 : 1834–1839.
12. FloTH, SmithKD, SatoS, RodriguezDJ, HolmesMA, et al. (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432 : 917–921.
13. RaffatelluM, BaumlerAJ (2010) Salmonella's iron armor for battling the host and its microbiota. Gut microbes 1 : 70–72.
14. StecherB, RobbianiR, WalkerAW, WestendorfAM, BarthelM, et al. (2007) Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS biology 5 : 2177–2189.
15. FischbachMA, LinH, ZhouL, YuY, AbergelRJ, et al. (2006) The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Proceedings of the National Academy of Sciences of the United States of America 103 : 16502–16507.
16. HantkeK, NicholsonG, RabschW, WinkelmannG (2003) Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proceedings of the National Academy of Sciences of the United States of America 100 : 3677–3682.
17. KonturekPC, KaniaJ, KukharskyV, RaithelM, OckerM, et al. (2004) Implication of peroxisome proliferator-activated receptor gamma and proinflammatory cytokines in gastric carcinogenesis: link to Helicobacter pylori-infection. Journal of pharmacological sciences 96 : 134–143.
18. MahajanS, DkharHK, ChandraV, DaveS, NanduriR, et al. (2012) Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARgamma and TR4 for survival. Journal of immunology 188 : 5593–5603.
19. KunduP, DeR, PalI, MukhopadhyayAK, SahaDR, et al. (2011) Curcumin alleviates matrix metalloproteinase-3 and -9 activities during eradication of Helicobacter pylori infection in cultured cells and mice. PloS one 6: e16306.
20. AdachiM, KurotaniR, MorimuraK, ShahY, SanfordM, et al. (2006) Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut 55 : 1104–1113.
21. DreyerC, KreyG, KellerH, GivelF, HelftenbeinG, et al. (1992) Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 68 : 879–887.
22. MansenA, Guardiola-DiazH, RafterJ, BrantingC, GustafssonJA (1996) Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochemical and biophysical research communications 222 : 844–851.
23. RicoteM, LiAC, WillsonTM, KellyCJ, GlassCK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391 : 79–82.
24. WahliW, MichalikL (2012) PPARs at the crossroads of lipid signaling and inflammation. Trends in endocrinology and metabolism: TEM 23 : 351–363.
25. SuCG, WenX, BaileyST, JiangW, RangwalaSM, et al. (1999) A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. The Journal of clinical investigation 104 : 383–389.
26. YangXY, WangLH, ChenT, HodgeDR, ResauJH, et al. (2000) Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. The Journal of biological chemistry 275 : 4541–4544.
27. Bassaganya-RieraJ, ReynoldsK, Martino-CattS, CuiY, HennighausenL, et al. (2004) Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 127 : 777–791.
28. DesreumauxP, DubuquoyL, NuttenS, PeuchmaurM, EnglaroW, et al. (2001) Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. The Journal of experimental medicine 193 : 827–838.
29. DubuquoyL, DharancyS, NuttenS, PetterssonS, AuwerxJ, et al. (2002) Role of peroxisome proliferator-activated receptor gamma and retinoid X receptor heterodimer in hepatogastroenterological diseases. Lancet 360 : 1410–1418.
30. KatayamaK, WadaK, NakajimaA, MizuguchiH, HayakawaT, et al. (2003) A novel PPAR gamma gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology 124 : 1315–1324.
31. LytleC, TodTJ, VoKT, LeeJW, AtkinsonRD, et al. (2005) The peroxisome proliferator-activated receptor gamma ligand rosiglitazone delays the onset of inflammatory bowel disease in mice with interleukin 10 deficiency. Inflammatory bowel diseases 11 : 231–243.
32. NakajimaA, WadaK, MikiH, KubotaN, NakajimaN, et al. (2001) Endogenous PPAR gamma mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology 120 : 460–469.
33. SaubermannLJ, NakajimaA, WadaK, ZhaoS, TerauchiY, et al. (2002) Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflammatory bowel diseases 8 : 330–339.
34. NecelaBM, SuW, ThompsonEA (2008) Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor gamma and nuclear factor-kappaB in macrophages. Immunology 125 : 344–358.
35. AreA, AronssonL, WangS, GreiciusG, LeeYK, et al. (2008) Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proceedings of the National Academy of Sciences of the United States of America 105 : 1943–1948.
36. KellyD, CampbellJI, KingTP, GrantG, JanssonEA, et al. (2004) Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nature immunology 5 : 104–112.
37. ZhengY, ValdezPA, DanilenkoDM, HuY, SaSM, et al. (2008) Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine 14 : 282–289.
38. BergerT, CheungCC, EliaAJ, MakTW (2010) Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proceedings of the National Academy of Sciences of the United States of America 107 : 2995–3000.
39. LengX, DingT, LinH, WangY, HuL, et al. (2009) Inhibition of lipocalin 2 impairs breast tumorigenesis and metastasis. Cancer research 69 : 8579–8584.
40. DubuquoyL, JanssonEA, DeebS, RakotobeS, KarouiM, et al. (2003) Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 124 : 1265–1276.
41. FlorinL, HummerichL, DittrichBT, KokocinskiF, WrobelG, et al. (2004) Identification of novel AP-1 target genes in fibroblasts regulated during cutaneous wound healing. Oncogene 23 : 7005–7017.
42. LiC, ChanYR (2011) Lipocalin 2 regulation and its complex role in inflammation and cancer. Cytokine 56 : 435–441.
43. KunduP, MukhopadhyayAK, PatraR, BanerjeeA, BergDE, et al. (2006) Cag pathogenicity island-independent up-regulation of matrix metalloproteinases-9 and -2 secretion and expression in mice by Helicobacter pylori infection. The Journal of biological chemistry 281 : 34651–34662.
44. CastanedaFE, WaliaB, Vijay-KumarM, PatelNR, RoserS, et al. (2005) Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology 129 : 1991–2008.
45. GargP, Vijay-KumarM, WangL, GewirtzAT, MerlinD, et al. (2009) Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. American journal of physiology Gastrointestinal and liver physiology 296: G175–184.
46. BrinckerhoffCE, MatrisianLM (2002) Matrix metalloproteinases: a tail of a frog that became a prince. Nature reviews Molecular cell biology 3 : 207–214.
47. HoisethSK, StockerBA (1981) Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291 : 238–239.
48. ImaiT, TakakuwaR, MarchandS, DentzE, BornertJM, et al. (2004) Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proceedings of the National Academy of Sciences of the United States of America 101 : 4543–4547.
49. WlodarskaM, WillingB, KeeneyKM, MenendezA, BergstromKS, et al. (2011) Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infection and Immunity 79 : 1536–1545.
50. SchreiberE, MatthiasP, MullerMM, SchaffnerW (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic acids research 17 : 6419.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 1
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
Najčítanejšie v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites