
Bartov syndróm asociovaný s epidermolysis bullosa junctionalis a s atréziou pyloru. Nekroptická kazuistika
Bart´s syndrome associated with epidermolysis bullosa junctionalis and with pyloric atresia. An autopsy case report
Bart‘s syndrome, in literature also known under the name CLAS (Congenital Localised Absence of Skin), first described by Bart in 1966 as congenital localized absence of skin, epidermolysis bullosa congenita and nail abnormalities. The authors present a macroscopic and histological findings of a newborn with Bart‘s syndrome, with epidermolysis bullosa junctionalis and atresia pylori, who died 17 days after birth and 13 days after surgery for pyloric stenosis.
Key words:
Bart´s syndrome – atresia pylori – epidermolysis bullosa junctionalis – CLAS
Autori:
Katarína Adamicová 1; Tomáš Balhárek 1; Lucia Lúčanová 2; Oľga Nyitrayová 3; Želmíra Fetisovová 4
Pôsobisko autorov:
Ústav patologickej anatómie JLF UK a UN Martin
1; Neonatologická klinika JLF UK a UN Martin
2; Cytopathos, spol. s r. o., Bratislava
3; Dermatovenerologická klinika JLF UK a UN Martin
4
Vyšlo v časopise:
Čes.-slov. Patol., 50, 2014, No. 4, p. 155-158
Kategória:
Původní práce
Súhrn
Bartov syndróm, v literatúre známy aj pod akronymickým názvom CLAS (Congenital Localised Absence of Skin), prvý raz opísal Bart v roku 1966 ako vrodené lokalizované chýbanie kože, epidermolysis bullosa congenita a nechtové abnormity. V príspevku autori prezentujú makroskopický aj histologický nález nekroptického prípadu novorodenca s Bartovým syndrómom, s epidermolysis bullosa junctionalis a s atréziou pyloru, ktorý zomrel po 17 dňoch od narodenia a 13 dňoch od operácie stenózy pyloru.
Kľúčové slová:
Bartov syndróm – atrézia pyloru – epidermolysis bullosa junctionalis – CLAS
Bartov syndróm (BS) je patologická kombinácia tkanivových zmien, ktorá sa prejavuje bulóznou chorobou kože typu epidermolysis bullosa (EB), nechtovými abnormitami a najmä lokalizovaným chýbaním rozlične veľkých ostrovčekov kože. V literatúre sa uvádza ako extrémne zriedkavé ochorenie (1). Ide o autozómovo dominantnú chorobu, ktorej podstatou je mutácia v exóne 73 COL7A1, spôsobujúca u postihnutých jednotlivcov nahradenie arginínu glycínom v „triple-helical domain“ kolagénu typu VII.
OPIS PRÍPADU
Klinický nález
Išlo o chlapca z prvej gravidity, rodinná anamnéza bola bez pozoruhodností. Začiatok prenatálnej diagnostiky bol od 10. týždňa gravidity, matka bola vyšetrená šesť ráz. Priebeh gravidity bol do 34. týždňa bez komplikácií. Náhle bola diagnostikovaná parciálna abrupcia placenty a vykonané vybavenie plodu per sectionem caesaream. Popôrodná adaptácia novorodenca bola dobrá, Apgarovej skóre 10/10/10 b, pôrodná hmotnosť 2 000 g, dĺžka 44 cm. Na pravej dolnej končatine bol prítomný rozsiahly defekt kože od kolena až na dorsum pedis.
Okrem najväčšieho defektu sa postupne na fragilnej a tenkej koži v rozličnej lokalizácii vytvárali pľuzgiere a plochy rán, najmä po manipulácii s dieťaťom. Kožný nález bol predbežne uzavretý ako EB. Na nechtoch dolných končatín boli zaznamenané dystrofické zmeny ako praskliny, strata transparentnosti nechtovej platničky a jej škvrnitosť.
Ďalšími znázorňovacími a klinickými vyšetreniami sa zistil kompenzovaný ventrikuloseptálny defekt srdca a atrézia pyloru. Na štvrtý deň života sa realizovala laparotómia s resekciou atretickej časti pyloru a s následnou anastomózou duodena. Výkon bol ukončený revíziou operačnej oblasti. Pooperačne dieťa vyžadovalo umelú pľúcnu ventiláciu. Postupne bola zistená pozitívna hemokultúra na dva druhy stafylokokov a progresia bolestivých reakcií dieťaťa. Na deviaty pooperačný deň sa stav pacienta zhoršil, pri minimálnej manipulácii reagoval spazmom a významnou desaturáciou. Napriek kontinuálnej resuscitačnej liečbe nastúpilo multiorgánové zlyhanie a rozvrat vnútorného prostredia. Vo veku 17 dní dieťaťa ošetrujúci lekár konštatoval exitus letalis.
Nekroptický nález
Koža bola na celom tele fragilná, poškodzovala sa aj pri najjemnejšej manipulácii. Na končatinách, chrbte a hlave boli početné povrchové kožné defekty so živočervenou, červenou a žltkastou spodinou (obr. 1). Niekde v okrajoch sa vyskytovali porušené pľuzgiere až buly. Na pravej dolnej končatine od dolnej časti stehna až po stupaj chýbal kožný kryt (obr. 2).


Na bruchu bola prítomná operačná rana s dĺžkou 7 cm s krustou, bez dehiscencie. Nechty boli dystrofické, so stratou translucencie, žltkasté, s pozdĺžnymi ryhami.
Vnútornou obhliadkou sa potvrdil klinicky zistený septálny defekt s priemerom 2 mm. Foramen ovale bolo otvorené, ductus arteriosus kontrahovaný. Ostatný nález bol primeraný veku.
Stav po anastomóze pyloru pre atréziu bol v norme hojenia, bez dehiscencie sutúry a bez iných zmien na sliznici (obr. 3).

Histologický nález excízií z kože
Vo všetkých vzorkách išlo o kožu novorodenca na hranici zrelosti. V niektorých lezionálnych excíziách kompletne absentovala epiderma (obr. 4). V periférnych častiach denudácie, v oblasti dermálno-epidermovej junkcie (DEJ) boli pozorované štrbiny bez celulizácie („cell-free“). Kórium bolo zväčša bez výraznejšej zápalovej celulizácie (obr. 5). Obsahovalo adnexálne štruktúry vrátane velusových vlasových folikulov. Excízie z oblasti pľuzgierov mali štrbiny v oblasti DEJ, bez výraznejšej celulizácie obsahu alebo okolitého tkaniva.
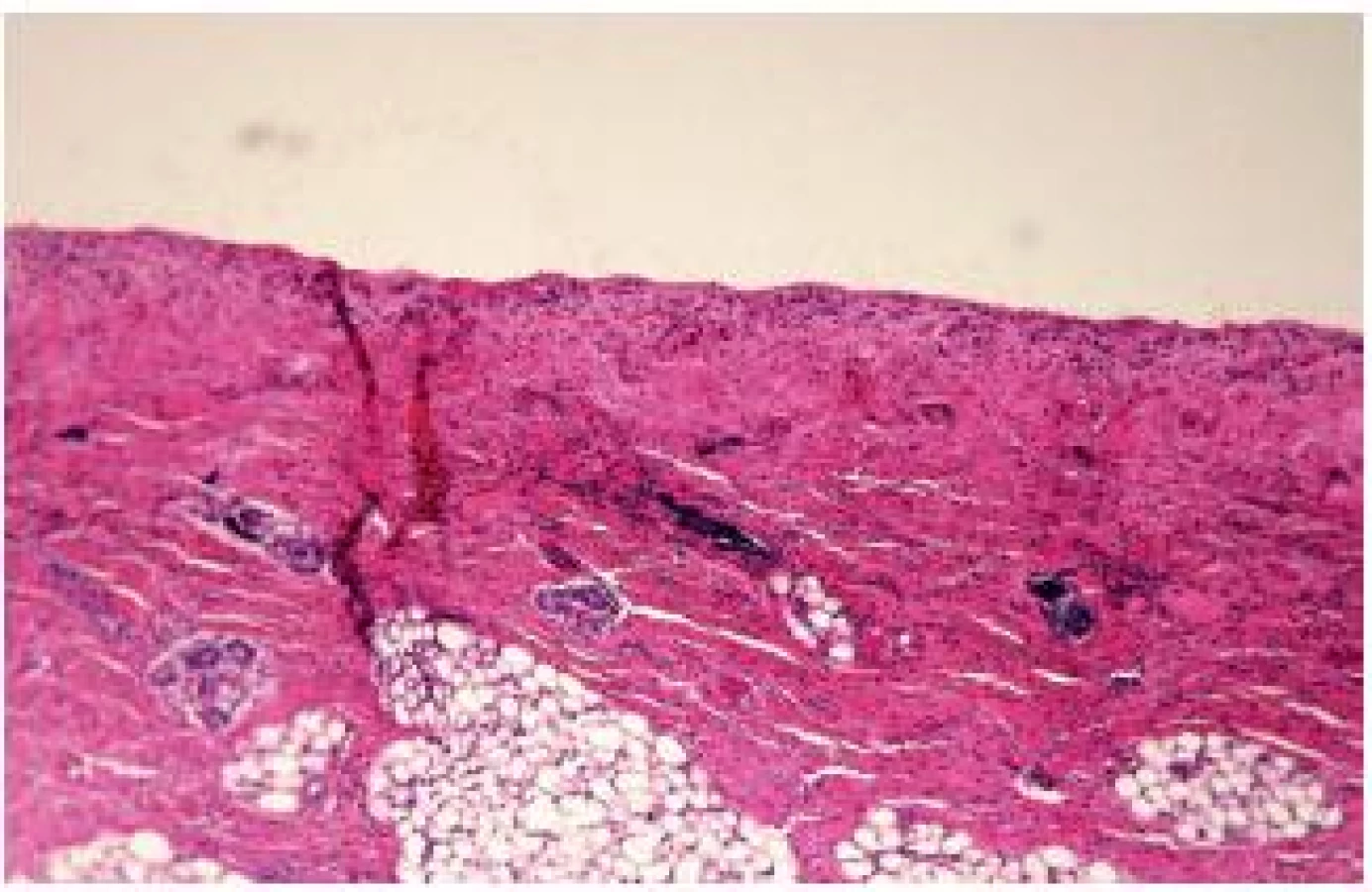

Aj napriek nedostatočnej kvalite materiálu pochádzajúceho z nekropsie sme sa pokúsili o imunohistochemický dôkaz laminínu a kolagénu IV. S rezervou bolo možné nález pokladať za typický, s prítomnosťou kolagénu IV antigénov na spodine pľuzgiera (obr. 6) a chýbaním laminínu v bazálnych bunkách epidermy.

Detailnejšie bolo možné pozorovať pľuzgier v polotenkých rezoch (obr. 7), ako aj absenciu hemidesmozómov a subbazálnych denzných platničiek v oblasti lamina lucida elektrónovomikroskopickým vyšetrením (obr. 8).


Časť I. a II. nekroptickej záverečnej diagnózy
I.:
- Mors neonati
- Partus praeterminalis operativus per sectionem caesaream in hebdomade XXXIV. propter abruptionem placentae praecocem (P03.4, P03.4)
- Immaturitas neonati (P07.3)
- Epidermolysis bullosa congenita (typus junctionalis) (Q81.9)
- Atrophia cutis congenita reg. membri infer. l. dx. („Congenital Localized Absence of Skin – CLAS“)
- Atresia pylori congenita – st. post operationem (Q40.8)
- Sepsis neonati – clinici scriptu (P36.8)
II.:
- Dysfunctio organorum internorum multiplex („Multiple Organ Dysfunction Syndrome – MODS“) (R57.8)
Diskusia
Bartov syndróm sa pokladá za nešpecifický prejav epidermolysis bullosa, na potvrdenie ktorej sa vyžadujú špeciálne vyšetrenia, vrátane molekulovogenetických. Ide o eponym pre tri klinické patologické zmeny, a to vrodené lokalizované chýbanie kože (CLAS – Congenital Localised Absence of Skin), epidermolysis bullosa (EB) a nechtové aberácie či abnormality. CLAS sa v súčasnosti pokladá za jednu z manifestácií EB, a to nielen dystrofického typu, ale aj junkčnej EB, najmä typov spojených s atréziou pyloru (2).
Ako najčastejšie vysvetlenie pre vznik vrodenej absencie kože sa v literatúre uvádza možná mechanická lézia spôsobená intrauterinnou fyzikálnou traumou (3). Táto teória prichádza do úvahy najmä vtedy, keď ide o jednostranné lézie. V prípade obojstranných a symetrických lézií kože je skôr akceptovaná teória McKinstera a spol. (4), ktorí vysvetľujú túto poruchu vývinovo, poruchou diferenciácie kože v priebehu Blaschkových línií. V prípade pacienta z horeuvedenej kazuistiky išlo o jednostranné chýbanie kože na pravej dolnej končatine, čo môže podporovať patogenézu intrauterinnej traumatizácie.
Napriek tomu, že molekulová báza BS nie je známa vo všetkých detailoch, je dokázané, že syndróm sa prejavuje autozómovo dominantnou dedičnosťou s kompletnou penetráciou a variabilnou expresiou (3), čo ako prvý dokázal Bart a spol. vyšetrením 26 členov jednej rodiny s výskytom kompletnej penetrácie ochorenia (5). U pacientov bola zaznamenaná absencia kože na dolných končatinách, pľuzgiernaté ochorenie kože aj slizníc a defekty nechtov. Sú však známe aj prípady, v ktorých nie všetci členovia rodiny s EB sú postihnutí aj BS, a naopak, nie všetci pacienti s BS musia mať diagnostikovanú EB. Bartov syndróm môže byť asociovaný s akoukoľvek formou EB, najčastejšie sa však opisuje výskyt dystrofickej EB (DEB) (6). V čase Bartových vyšetrení nebolo k dispozícii ultraštrukturálne ani imunohistochemické vyšetrenie, preto sa o jednoznačnom subtype EB môže iba uvažovať, aj keď neskôr Zelickson a spol. dokázali v materiáli z originálnych prípadov vyšetrovaných Bartom dominantnú DEB (7).
V prípade, ktorý je predmetom našej kazuistiky, bol nález uzatvorený ako zriedkavé bulózne ochorenie, bez možnosti potvrdiť spôsob dedičnosti, najskôr typu EB junctionalis (EBJ). Diagnostická istota bola skupinovo podoprená elektrónovomikroskopickým vyšetrením, charakterizovaným hypopláziou, resp. až chýbaním hemidesmozómov, s defektmi, alebo chýbaním subbazálnych denzných platničiek v lamina lucida (8), ďalej vyšetrením polotenkých rezov, ktoré zdôraznili tvorbu subepidermálnych pľuzgierov bez významnejšej zápalovej odozvy, ako aj prítomnosťou atrézie pyloru, ktorá sa uvádza najmä v skupine EBJ, a klinickým obrazom (9).
Atrézia pyloru v spojení s EB je zapríčinená mutáciou ITGA6, ITGB4 a PLEC génov. Tieto gény sú zodpovedné za tvorbu proteínov potrebných v koži a digestívnom systéme. Mutácia ITGB4 je najčastejšia, je opísaná v 80 % prípadov. Mutácia ITGA6 je prítomná v 5 % prípadov. Tieto gény formujú proteín označovaný ako α6β4 integrín. Tento proteín má dôležitú úlohu v pripevnení a stabilizácii epidermy k hlbším častiam kože. Jeho chýbanie spôsobuje fragilitu kože so všetkými ďalšími klinickými a patologickými následkami.
Približne 15 % prípadov EB v spojení s pylorickou atréziou má dokázanú mutáciu PLEC génu. Tento gén je zodpovedný za tvorbu proteínu plektínu. Podobne ako α6β4 integrín plektín pomáha upevniť epidermis na hlbšie časti kože a jeho defektná tvorba býva príčinou separácie v mieste bazálnej membrány (10).
✉ Adresa pro korespondenci:
Prof. MUDr. Katarína Adamicová, PhD.
Ústav patologickej anatómie JLF UK a UNM
Kollárova 2, 036 59 Martin
e-mail: adamicova@jfmed.uniba.sk
mobil: +421 903 513 122
Zdroje
1. Rajpal A, Mishra R, Hajirnis K, Shah M, Nagpur N. Bart‘s syndrome. Indian J Dermatol 2008; 53(2): 88-90.
2. Narter F, Büyükbabani N, Yrarli H, öztürk S, Ergüven M. Bart´s syndrome associated with pyloric and choanal atresia. The Turkish J of Pediatrics 2013; 55(2): 214-217.
3. Pereira de Almeid NA, Serafini F, Marchiori J, Del Moro JG. Do you know this syndrome? An Bras Dermatol 2010; 85(1): 119-121.
4. Duran-McKinster C, Rivera-Franco A, Tamayo L, de la Luz Orozco-Covarrubias M, Ruiz-Maldonado R. Bart syndrome: the congenital localized absence of skin may follow the lines of Blaschko. Report of six cases. Pediatr Dermatol 2000; 17(3): 179-182.
5. Smith SZ, Cram DL. A mechanobullous disease of the newborn. Bart‘s syndrome. Arch Dermatol 1978; 114(1): 81-84.
6. Bajaj DR, Quereshi A. Bart´s syndrome a case report. J of Pakist Assoc of Dermatol 2008; 18 : 113-115.
7. Zelickson B, Matsumura K, Kist D, Epstein EH Jr, Bart BJ. Bart´s syndrome: Ultrastructure and genetic linkage. Arch Dermatol 1995; 131(6): 663-668.
8. Bučková H, Buček J. Epidermolysis bullosa congenita. Brno: IDVZP; 2000 : 3-122.
9. Calonje E, Brenn T, Lazar A, McKee PH, eds. McKee´s Pathology of the skin. (4th edn). Philadelphia: Elsevier; 2012 : 938-939.
10. Pfendner E, Uitto P. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol 2005 : 124(1): 111-115.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2014 Číslo 4
Najčítanejšie v tomto čísle
- Postavenie a význam cytológie moča v diagnostike uroteliálnych nádorov
- Bartov syndróm asociovaný s epidermolysis bullosa junctionalis a s atréziou pyloru. Nekroptická kazuistika
- International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia 2012
- Novinky v patologii prostaty