
WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition
WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition
In early 2013, the new classification of tumours of soft tissue and bones was released. This edition belongs to the fourth series of so-called Blue Books published under the auspices of the World Health Organisation (WHO). The current classification follows the previous third edition, from which it differs in several aspects. The vast majority of changes are related to the soft tissue tumour section, which was enriched with three new chapters, some entities or terms were removed, new diagnoses were introduced, and several tumours were reallocated to other categories. Albeit to a lesser extent, similar changes have occurred also in the classification of bone tumours. Compared to the previous edition, more detailed molecular and cytogenetic data were incorporated in the current issue. The rapidly increasing knowledge of the genetics of mesenchymal tumours allows us to make more accurate diagnoses as well as to better understand of the pathogenesis of these lesions. However, abundant molecular and cytogenetic data highlight an increasing problem of growing numbers of genetic overlaps even among quite different tumours. The coexistence of several grading systems of soft tissue tumours is another controversial issue mentioned in the recent WHO classification. The main advantages and limitations of the two most widely used grading systems are also discussed.
Keywords:
WHO classification – sarcoma – soft tissues – bones – tumour
Autoři:
Iva Zambo; Karel Veselý
Působiště autorů:
I. patologicko-anatomický ústav LF MU a FN u sv. Anny v Brně
Vyšlo v časopise:
Čes.-slov. Patol., 50, 2014, No. 2, p. 64-70
Kategorie:
Přehledový článek
Souhrn
Na začátku roku 2013 vyšla nová klasifikace tumorů měkkých tkání a kostí, která patří mezi 4. sérii tzv. „Modrých knih“ vydávaných pod záštitou Světové zdravotnické organizace (WHO). Tato klasifikace plynule navazuje na předchozí třetí vydání, od něhož se liší v několika aspektech. Převážná většina změn se týká sekce měkkotkáňových tumorů, která byla nově obohacena o tři kapitoly, došlo k odstranění některých jednotek, popřípadě termínů, představení nových diagnóz a přesunu několika jednotek pod odlišné kategorie nádorů. K obdobným změnám, i když menšího rozsahu, došlo i v klasifikaci kostních tumorů. Oproti předchozí edici byla v současném vydání zahrnuta detailnější molekulární a cytogenetická data. Rychle rostoucí znalost genetiky mezenchymálních tumorů nám umožňuje stanovit přesnější diagnózu, i lépe pochopit patogenezi těchto lézí. Množství molekulárních a cytogenetických dat ale poukazuje na rostoucí problém stále početnějších sdílených genetických alterací i mezi zcela odlišně se chovajícími tumory. Dalším sporným tématem, zmíněným v recentní WHO klasifikaci, je paralelní existence více gradingových systémů měkkotkáňových tumorů. Jsou diskutovány i hlavní výhody a limitace dvou nejvíce používaných gradingových systémů.
Klíčová slova:
WHO klasifikace – sarkomy – měkké tkáně – kosti – tumor
WHO klasifikace tumorů měkkých tkání a kostí doznala v posledních desetiletích výrazných změn. Většina těch nejpodstatnějších byla představena již ve 3. vydání, které vyšlo na konci roku 2002 (1). K podstatným pokrokům v koncepci měkkotkáňových tumorů patří zjištění, že některé morfologicky jednoznačně benigní léze (např. benigní fibrózní histiocytom či difúzní typ obrovskobuněčného tumoru šlachové pochvy) mohou velice vzácně vzdáleně metastazovat, a tuto skutečnost nelze předpovědět na základě morfologického či jiného vyšetření.
Kriticky byla v předchozím vydání diskutována diagnóza tzv. maligního fibrózního histiocytomu (MFH), synonymicky označovaného jako nediferencovaný pleomorfní sarkom. Většinu pleomorfních sarkomů lze totiž při pečlivém vyšetření dále subklasifikovat a tumor správně zařadit jako pleomorfní leiomosarkom, high-grade myxofibrosarkom, dediferencovaný či pleomorfní liposarkom nebo pleomorfní rabdomyosarkom. Tzv. pleomorfní MFH byl v minulosti nejčastěji diagnostikovaným sarkomem u dospělých pacientů, při důsledné aplikaci kritérií WHO klasifikace by však jeho četnost neměla překročit 5 % zhoubných měkkotkáňových tumorů (2). Ve 4. vydání je od zastaralého názvu MFH zcela upuštěno, a tyto léze jsou nyní zařazeny do nové samostatné kategorie nediferencovaných / neklasifikovaných sarkomů (3), o kterých bude podrobněji pojednáno níže.
Hemangiopericytom nevykazuje známky pericytární diferenciace, proto byl z kategorie pericytických (perivaskulárních) tumorů přeřazen mezi fibroblastické / myofibroblastické nádory, přesněji k extrapleurálnímu solitárnímu fibróznímu tumoru. Z recentní klasifikace byl termín hemangiopericytom odstraněn úplně.
U řady tumorů není známa linie diferenciace, kterou tyto léze rekapitulují, a to i v případech s jasně definovanou morfologií, a často i dobře specifikovaným imunofenotypem. Kategorie nádorů s nejistou diferenciací se tak postupně rozrůstá o tumory původně řazené do jiné skupiny nebo o nově uvedené jednotky (2,4).
Nedílnou součástí obou posledních WHO klasifikací mezenchymálních tumorů je rychle rostoucí množství genetických dat, která zpřesňují diagnózu. V současnosti patří detekce specifických chromozomálních alterací u některých tumorů (např. synoviálního sarkomu, Ewingova sarkomu) do standardního diagnostického algoritmu a finální diagnóza těchto lézí by měla být stanovena až po molekulárně genetickém vyšetření (in situ hybridizaci, polymerázové řetězové reakci) verifikujícím přítomnost sledované přestavby genu. Na druhou stranu přibývá stále více jednotek, u kterých se vyskytují stejné genetické změny, což může být v některých případech zavádějící. Sdílené chromozomální aberace byly identifikovány u nádorů s podobnou morfologií (např. u vřetenobuněčného lipomu a celulárního angiofibromu), dále u tumorů s hybridní morfologií (u dermatofibrosarcoma protuberans a obrovskobuněčného fibroblastomu), ale i u nádorů histologicky zcela odlišných (např. světlobuněčného sarkomu a angiomatoidního fibrózního histiocytomu). Identické genetické alterace byly pozorovány i u tumorů vycházejících z různých zárodečných listů, např. u infantilního fibrosarkomu a sekretorického karcinomu (4).
Histologický grade sarkomů měkkých tkání je považován za nejvýznamnější prognostický faktor. Dosud však neexistuje jednotné, univerzálně akceptované gradingové schéma. Mezi v současnosti nejvíce užívaná schémata patří dva třístupňové, tzv. NCI (National Cancer Institute) a FNCLCC (Fédération Natinale des Centres de Lutte Contre la Cancer) gradingové systémy. Oba jsou založené na kvantifikaci mitotické aktivity a rozsahu koagulační nekrózy, jejich hlavní komponentou je histologický typ tumoru. FNCLCC gradingový systém je přesněji definovaný a u dospělých pacientů by měl být preferovaný (5). Hlavními slabinami obou schémat je správné určení histologického typu, případně subtypu sarkomu, sporně aplikovatelná kritéria počtu mitotických figur u hypocelulárních (např. myxoidních) tumorů a stanovení rozsahu intralezionální nekrózy u objemných tumorů, z nichž je histologicky zpracována jen malá část a makroskopický odhad rozsahu nekrózy může být klamný. Velmi sporné je rovněž hodnocení nekrózy u nádorové tkáně resekované po předchozí chemoterapii, případně radioterapii. U dediferencovaného a hypercelulárního myxoidního liposarkomu, rabdomyosarkomu, Ewingova sarkomu, alveolárního sarkomu měkkých částí, epiteloidního sarkomu, světlobuněčného sarkomu měkkých tkání, extraskeletárního myxoidního chondrosarkomu a u tumorů vzácně metastazujících, je grading méně informativní než samotný typ tumoru (5). V nejnovější WHO klasifikaci je představen tzv. molekulární gradingový systém založený na identifikaci expresního profilu celkem 67 genů souvisejících s komplexitou chromozomů a řídících mitózy (CINSARC, Complexity Index in SARComas), který byl popsán na velkém souboru sarkomů s komplexním genomovým profilem. Tento molekulární gradingový systém u high-grade sarkomů, gastrointestinálních stromálních tumorů (GIST), ale např. i u karcinomu mammy a difúzního velkobuněčného B-lymfomu prognosticky překonává histologický grading (6,7).
Stejně jako v předchozím vydání, je i v recentní WHO klasifikaci definován biologický potenciál mezenchymálních nádorů do tří základních kategorií, na nádory benigní, intermediární a maligní.
Benigní nádory většinou lokálně nerecidivují, jejich případné rekurence rostou nedestruktivně a řešeny jsou kompletní excizí. V extrémně vzácných případech může však i benigní tumor vzdáleně metastazovat, což bylo dobře dokumentováno u již zmíněného benigního fibrózního histiocytomu nebo difúzního typu obrovskobuněčného tumoru šlachové pochvy (8). Metastatický potenciál těchto nádorů nelze nijak předpovědět na základě morfologického histopatologického vyšetření.
Tumory s intermediárním biologickým chováním zahrnují dvě separátní podkategorie: nádory lokálně agresivní a tumory zřídka metastazující. Lokálně agresivní intermediární tumory se vyznačují tendencí k častým místním recidivám, které rostou infiltrativně a lokálně destruktivně, nemají však metastatický potenciál. Mezi typické zástupce této kategorie patří desmoidní fibromatóza či např. osteoblastom. Pro zřídka metastazující intermediární tumory je také charakteristický lokálně agresivní růst. V méně než 2 % případů však mohou zakládat vzdálené metastázy, a toto riziko nelze spolehlivě předpovědět na základě histologického (či jiného) vyšetření. Prototypem vzácně metastazujících tumorů je plexiformní fibrohistiocytární tumor, angiomatoidní fibrózní histiocytom nebo chondroblastom. Pro zajištění lokální kontroly intermediárních tumorů je nezbytná široká excize s dostatečným bezpečnostním lemem zdravé tkáně.
Maligní nádory měkkých tkání, tedy sarkomy, mohou růst destruktivně a kromě tendence k lokálním recidivám mají i nezanedbatelné riziko zakládání vzdálených metastáz, které se pohybuje v rozmezí od 20 do téměř 100 % v závislosti na histologickém typu a gradu. Některé morfologicky low-grade sarkomy metastazují pouze ve 2 – 10 %, ale v lokálních recidivách může jejich histologický grade progredovat do vyšších stupňů, a s ním i riziko vzdálené diseminace. Typickými zástupci jsou např. myxofibrosarkom či leiomyosarkom (1,2).
ZMĚNY V KLASIFIKACI MĚKKOTKÁŇOVÝCH SARKOMŮ
Sekce měkkotkáňových tumorů je nově obohacena o tři kapitoly: gastrointestinální stromální tumory, tumory z nervových obalů a nediferencované / neklasifikované sarkomy (tab. 1). Do poslední jmenované kategorie byla přesunuta skupina nediferencovaných pleomorfních sarkomů (dříve MFH) původně zařazených mezi tzv. fibrohistiocytární tumory. Pojem MFH tak přestal existovat.

Na základě detailnějšího poznání histogeneze došlo k malým přesunům některých benigních nádorů mezi jednotlivými kapitolami, lépe jsou např. definovány pericytární (perivaskulární) tumory. V nové WHO klasifikaci byl odstraněn termín hemangiopericytom a vypuštěna diagnóza smíšeného liposarkomu.
Bylo definováno i několik nových nozologických jednotek, které jsou představovány především raritními nádory. Patří mezi ně pseudomyogenní hemangioendoteliom (neboli epiteloidnímu sarkomu podobný hemangioendoteliom) zařazený mezi cévními nádory. Kapitola tumorů nejisté diferenciace se rozrostla o 4 jednotky: akrální fibromyxom, atypický fibroxantom a dva nově popsané nádory, kterými jsou hemosiderotický fibrolipomatózní tumor a fosfaturický mezenchymální tumor. Nově byla také definována kategorie hybridních nádorů nervové pochvy. Nové vydání WHO klasifikace je rozšířeno o molekulárně genetická data celé řady jednotek (4).
Adipocytární tumory
V této kategorii došlo k několika změnám. Atypický lipomatózní tumor (ALT) je při lokalizaci v retroperitoneu, mediastinu a oblasti semenného provazce označován za dobře diferencovaný liposarkom. Tyto tumory rostou lokálně agresivně, ale nikdy nemetastazují, proto jim byl přiřazen nový morfologický kód MKN-O 8850/1, místo původního 8851/3. V chirurgicky obtížně přístupných oblastech, kde není splněn předpoklad široké excize, jsou však tyto léze doprovázeny častými recidivami s rizikem dediferenciace, a poté i schopností vzdáleně metastazovat. Nově bylo upřesněno, že high-grade komponenta v dediferencovaném liposarkomu může připomínat pleomorfní liposarkom. Hovoří se o tzv. homologní lipoblastické diferenciaci. Pro verifikaci diagnózy ALT (resp. dobře diferencovaného liposarkomu) i dediferencovaného liposarkomu lze užít detekci některého z amplifikovaných genů, např. MDM2 (12q15) nebo CDK4 (12q14.1), a to buď molekulárně geneticky nebo pomocí imunohistochemie na úrovni proteinu.
Ve 4. vydání byl vypuštěn termín kulatobuněčný liposarkom, včetně MKN-O kódu 8853/3. Jedná se o high-grade myxoidní liposarkom morfologicky charakterizovaný přítomností hypercelulární komponenty (kulatobuněčné nebo vřetenobuněčné) a definovaný reciproční translokací FUS-DDIT3 nebo v přibližně 5 % případů zastoupenou EWSR1-DDIT3 (obr. 1). Odstraněna byla také diagnóza smíšeného liposarkomu, který je pravděpodobně představován nádory s neobvyklými morfologickými projevy dediferenciace (viz dediferencovaný liposarkom s homologní lipoblastickou diferenciací). Zůstala kategorie „liposarkom, dále neklasifikovaný“, protože vzácné případy skutečně nelze spolehlivě zařadit pod hlavičku žádné známé jednotky.

Fibroblastické / myofibroblastické nádory
Důležitou změnou v této kategorii je poznání, že nodulární fasciitis a její varianty (proliferativní fasciitis a myositis) nejsou reaktivní léze, ale nádorové procesy charakterizované reciproční translokací t(17;22) s fúzí genů MYH9-USP6. Nově tedy byly opatřeny morfologickým kódem MKN-O 8828/0.
Kapitola byla rozšířena o dva nádory, původně uváděné ve WHO klasifikaci kožních tumorů. Obrovskobuněčný fibroblastom spadá do kategorie intermediárních lokálně agresivních neoplázií. Druhou, nově zařazenou jednotkou, je dermatofibrosarcoma protuberans, který velmi vzácně metastazuje, čemuž ale prakticky vždy předchází transformace do fibrosarkomatózní varianty. Vzhledem k extrémně nízké schopnosti zakládat vzdálené metastázy u myxoinflamatorního fibroblastického sarkomu, je v nové WHO klasifikaci pro tuto jednotku propagován alternativní název atypický myxoinflamatorní fibroblastický tumor.
K dalším změnám došlo v rámci jednotek zahrnovaných pod diagnózu extrapleurálního solitárního fibrózního tumoru (SFT). Odstraněn byl termín hemangiopericytom (obr. 2). Obrovskobuněčný angiofibrom je nyní považován za synonymické označení pro SFT.

V rámci nově prezentované koncepce maligních fibroblastických nádorů se předpokládá, že mezi low-grade fibromyxoidním sarkomem a sklerózujícím epiteloidním sarkomem zřejmě existuje blízký vztah, vyjádřený sdílením identických genetických přestaveb genu FUS, histologicky s hybridní morfologií u některých případů (4).
Tzv. „fibrohistiocytární“ tumory
Nejdůležitější, ale očekávanou změnou oproti 3. vydání, je definitivní odstranění v minulosti hojně užívané diagnózy tzv. MFH, neboli nediferencovaného pleomorfního sarkomu. Jednotky, které nelze za použití v současnosti dostupných technologií blíže klasifikovat, jsou nyní označovány jako nediferencované / neklasifikované sarkomy, a mezi měkkotkáňovými tumory tvoří samostatnou skupinu (obr. 3).

V neurčitě definované kategorii tzv. „fibrohistiocytárních“ tumorů tak zůstaly pouze obrovskobuněčný tumor šlachové pochvy (lokalizovaný či difúzní), hluboký benigní fibrózní histiocytom, plexiformní fibrohistiocytární tumor a obrovskobuněčný tumor měkkých tkání. Poslední dvě jmenované jednotky patří mezi intermediární, vzácně metastazující nádory.
Hladkosvalové tumory
V této kategorii došlo jen k malým změnám. Původně zde uváděný angioleiomyom byl přesunut mezi pericytické (perivaskulární) tumory. Na základě cytogenetické analýzy profilování genové exprese bylo identifikováno několik molekulárních subtypů leiomyosarkomu, včetně méně diferencovaných variant. S nimi patrně úzce souvisí některé tumory dříve klasifikované jako nediferencované pleomorfní sarkomy. Takové léze by mohly být označeny za dediferencovné leioyosarkomy (8).
Pericytické (perivaskulární) tumory
Jednotky zahrnuté v této kategorii vykazují jasné znaky myoidní diferenciace a typicky obkružují cévy. Imunohistochemicky u nich prokazujeme pozitivitu hladkosvalového aktinu, často také h-caldesmonu, exprese desminu je však negativní. Do této skupiny byly přesunuty myofibrom a myofibromatózy (v předchozím vydání řazené mezi fibroblastické / myofibroblastické tumory) a angioleiomyom, původně uváděný mezi hladkosvalovými nádory. Myopericytom, myofibrom (i myofibromatóza) a angioleiomyom představují morfologické kontinuum téhož nádoru.
Nádory příčně pruhovaného svalu
Jedinou důležitou změnou v této skupině tumorů bylo vyčlenění vřetenobuněčného rabdomyosarkomu, původně uvedeného jako podtyp embryonálního rabdomyosarkomu. Vřetenobuněčný a sklerózující rabdomyosarkom spolu pravděpodobně úzce souvisí, nesdílí žádnou genetickou alteraci s embryonálním ani alveolárním rabdomyosarkomem, a liší se i klinickou manifestací. Vřetenobuněčný typ bývá u dětí a adolescentů lokalizován paratestikulárně a má povzbudivou prognózu. U dospělých tato varianta rabdomyosarkomu postihuje zejména oblast hlavy a krku, chová se agresivněji a i vzhledem k nižší chemosenzitivitě má horší prognózu než v mladších věkových skupinách. Sklerózující rabdomyosarkom postihuje oblast hlavy a krku a končetiny ve všech věkových kategoriích, a jeho prognóza ještě není zcela jasná. Dále bylo upřesněno, že případy se smíšenou morfologií alveolárního a embryonálního rabdomyosarkomu většinou postrádají chimérický gen PAX3-FOXO1 a pravděpodobně se tedy jedná o variantu embryonálního rabdomyosarkomu, nikoli alveolárního, jak se původně předpokládalo.
Vaskulární tumory
Kategorie cévních tumorů byla obohacena o nově uvedený pseudomyogenní hemangioendoteliom, synonymicky označovaný jako epiteloidnímu sarkomu podobný hemangioendoteliom. Jedná se o intermediární, vzácně metastazující tumor, který je typicky lokalizován na končetinách adolescentů a mladých dospělých a ve více než polovině případů bývá multicentrický. Obvykle vyrůstá v kůži, v případě multicentrického výskytu však postihuje i podkoží, příčně pruhované svaly i kosti.
Chondrooseózní nádory
Výčet jednotek v této skupině tumorů zůstal beze změny. Kategorie je zastoupena pouze chondromem měkkých tkání a extraskeletálním osteosarkomem. Kdysi uváděný extraskeletální myxoidní chondrosarkom nevykazuje žádné zřetelné známky chrupavčité diferenciace, a proto byl již ve 3. vydání WHO klasifikace přeřazen mezi tumory nejisté diferenciace. Mezenchymální chondrosarkom častěji než měkké tkáně primárně postihuje kosti, proto je uveden v sekci kostních tumorů, mezi maligními chondrogenními tumory (1,3).
Gastrointestinální stromální tumory
Jsou nově zařazenou kapitolou v sekci měkkotkáňových nádorů. V prakticky identické podobě jsou současně uvedeny i v recentní WHO klasifikaci nádorů trávícího systému (9). V souvislosti s nádory měkkých tkání má význam zejména výskyt GISTů v retroperitoneu, mezenteriu a pánvi. V obou nových WHO klasifikacích je ale tématu extragastrointestinálních GISTů věnováno minimum prostoru s ohledem na jejich raritní výskyt. V příslušné kapitole je tedy pojednáno o GISTech v trávícím traktu. Na základě rozsáhlých studií Miettinena a jeho spolupracovníků byla u těchto tumorů dobře definována prognostická kritéria, která umožňují zhodnotit riziko agresivního chování GISTů (10). Dále byla popsána nová skupina GISTů charakterizovaná ztrátou funkce komplexu sukcinátdehydrogenázy (SDH). Tyto tumory jsou nyní známé jako SDH-deficientní GISTy, nebo také GISTy pediatrického typu, postrádají KIT i PDGFRA mutaci. Vyskytují se v řadě klinických variant a jejich biologické chování nekoreluje s počtem mitóz ani velikostí tumoru (9).
Nádory nervových obalů
Patří mezi nově začleněné kategorie ve svazku měkkých tkání a jsou tvořeny jednotkami původně uvedenými ve WHO klasifikacích tumorů centrálního nervového systému, nádorů hlavy a krku a kožních tumorů. V recentní WHO klasifikaci mezenchymálních nádorů byla poprvé představena kategorie hybridních tumorů nervové pochvy, které jsou reprezentovány kombinacemi hybridní schwannom/perineuriom, hybridní neurofibrom/schwannom, hybridní nádor z granulárních buněk/perineuriom a hybridní neurofibrom/perineuriom. Tyto nádory se chovají benigně, zřídka mohou lokálně recidivovat (4).
Nádory nejisté diferenciace
U nádorů zařazených do této kategorie není známa linie diferenciace, kterou tumory rekapitulují. Tyto léze jsou vymezeny morfologicky, v některých případech i imunohistochemicky nebo molekulárně-geneticky. Skupina nádorů nejisté diferenciace byla rozšířena o čtyři jednotky. Akrální (digitální) fibromyxom patří mezi benigní tumory dospělých pacientů, téměř vždy vyrůstá v blízkosti nehtového lůžka a je tvořený uniformními vřetenitými buňkami obklopenými hojnou myxoidní matrix.
Dalším, mezi mezenchymální neoplázie nově uvedeným nádorem, je atypický fibroxantom (AFX), který byl původně zařazen ve WHO klasifikaci kožních nádorů. AFX se v naprosté většině případů chová benigně, a to navzdory své „jednoznačně maligní“ morfologii. V extrémně vzácných případech může zakládat vzdálené metastázy.
Třetím, úplně poprvé uvedeným tumorem, je hemosiderotický fibrolipomatózní tumor (HFLT), někdy označovaný jako hemosiderotická fibrohistiocytární lipomatózní léze. Jedná se o vzácnou afekci postihující převážně dospělé, vyrůstající v podkoží na dorzu rukou a nohou. Léze je tvořená neostře ohraničenou proliferací zralé tukové tkáně a uniformních fibroblastů, s depozicí hemosiderinu. Až ve třetině případů lokálně recidivuje a má i potenciál k maligní progresi. V HFLT byla identifikována stejná translokace jako u myxoinflamatorního fibroblastického sarkomu (neboli atypického myxoinflamatorního fibroblastického tumoru), tedy t(1;10)(p22;q24). Obě jednotky se obdobně klinicky prezentují a v současnosti byly popsány případy se smíšenými morfologickými rysy.
Posledním, nově představeným nádorem, je fosfaturický mezenchymální tumor, který se objevuje u dospělých středního věku v libovolné lokalizaci a prostřednictvím fibroblastického růstového faktoru 23 vyvolává osteomalácii. Tyto léze jsou morfologicky tvořeny buňkami vzhledu a upořádání myofibroblastů či myopericytů, které jsou obklopeny hyalinizovanou matrix, místy s granulárními kalcifikacemi. Fosfaturický mezenchymální tumor je řazen do kategorie intermediárních, vzácně metastazujících nádorů (4).
V recentní klasifikaci již není pro Ewingův sarkom užíváno synonymického označení primitivní neuroektodermální tumor (PNET), a to z důvodů možné záměny s podobně označovanými lézemi v CNS a ženském genitálním traktu (obr. 4). Také je kriticky diskutována diagnóza maligního mezenchymomu, z výčtu jednotek opatřených morfologickým kódem MKN-O je vyřazen. Většina v minulosti popisovaných případů maligního mezenchymomu pravděpodobně představuje heterologní linii diferenciace v konkrétních sarkomech, nejčastěji v myxoidním liposarkomu, ALT nebo dediferencovaném liposarkomu (3).

Nediferencované / neklasifikované sarkomy
Jedná se o nově vzniklou skupinu maligních tumorů, která zahrnuje léze původně označované za tzv. MFH. Do této kategorie patří sarkomy, u kterých se žádnou dostupnou metodou nepodaří zjistit linii diferenciace. Jejich další subklasifikace je v současnosti založená na buněčné morfologii. Nyní tedy rozlišujeme 5 variant nediferencovaných sarkomů: nediferencovaný vřetenobuněčný, pleomorfní, kulatobuněčný a epiteloidní sarkom a nediferencovaný sarkom blíže nespecifikovaný. Přibližně čtvrtina nediferencovaných / neklasifikovaných sarkomů vzniká v souvislosti s předchozím ozářením.
Za pomocí molekulárně genetických metod byla odhalena celá řada chromozomálních přestaveb, převážně translokací, pomocí kterých jsou mezi nediferencovanými kulatobuněčnými sarkomy vyčleňovány některé genetické podskupiny. U části pediatrických pacientů byl např. identifikován fúzní gen CIC-DUX4, který kóduje chimerický protein deregulující geny podtřídy PEA3 rodiny ETS. Podobné geny jsou zvýšeně exprimované i v klasickém Ewingově sarkomu. Je tak vysvětlena morfologická podobnost kulatobuněčné varianty nediferencovaného sarkomu s Ewingovým sarkomem. Genetická data tumorů v této kategorii rychle přibývají. Lze očekávat, že v budoucnu dojde k reorganizaci celé kapitoly nediferencovaných / neklasifikovaných sarkomů.
ZMĚNY V KLASIFIKACI KOSTNÍCH NÁDORŮ
Obdobně jako v měkkotkáňové sekci, je v klasifikaci kostních nádorů samostatně vyčleněna kapitola nediferencovaného high-grade pleomorfního sarkomu, který byl ještě v předchozím 3. vydání označován jako MFH kostí a řazen mezi fibrohistiocytární kostní tumory (tab. 2). V některých kapitolách byl výčet primárních kostních lézí rozšířen o jednotky původně diskutované v jiných dílech WHO klasifikací (hematopoetické neoplázie, vaskulární tumory), také se rozrostla kapitola věnovaná nádorovým syndromům. Nově jsou uvedeny diagnózy ostechondromyxomu, obrovskobuněčné léze malých kostí a benigního notochordálního tumoru (3).
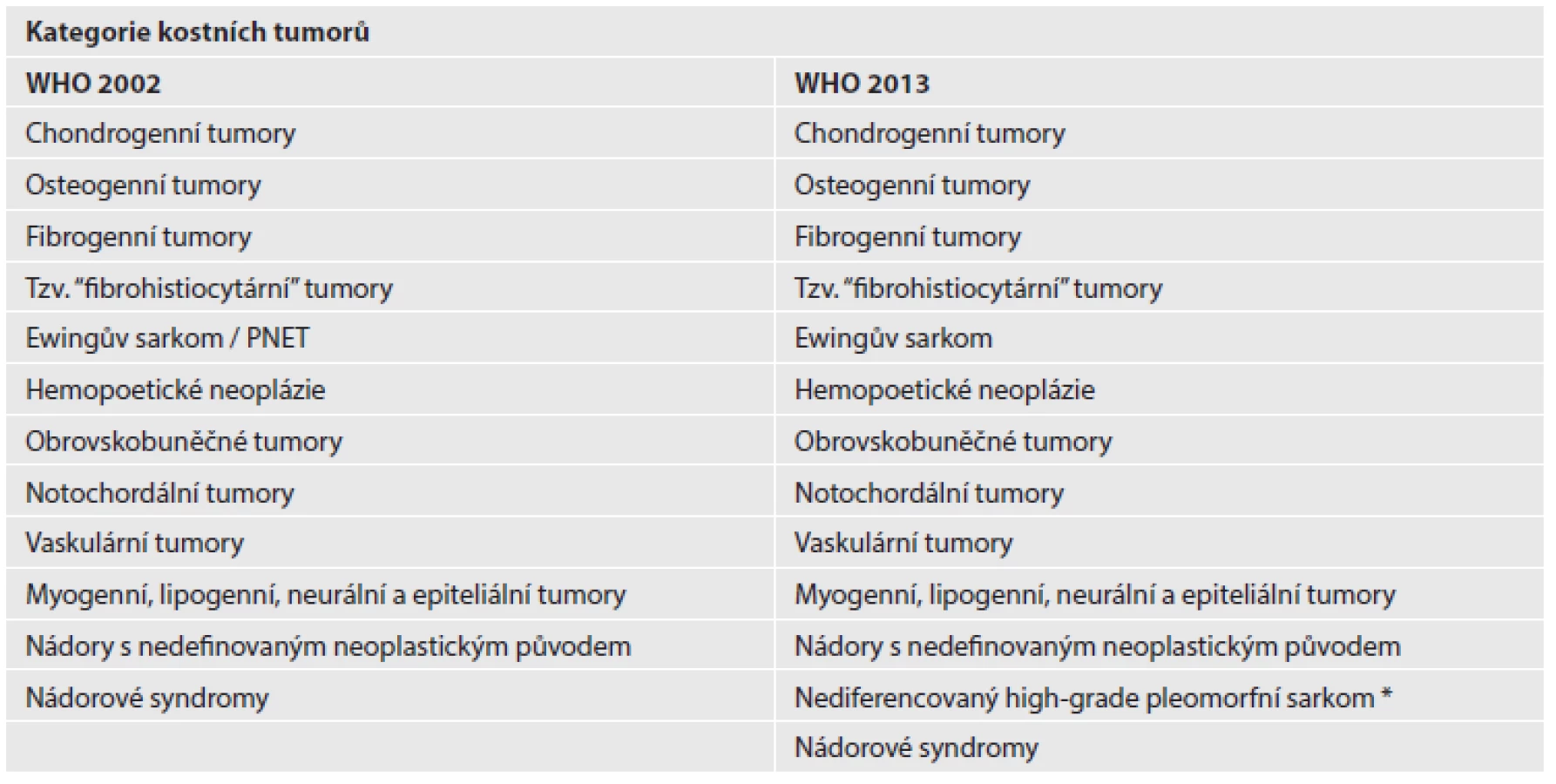
Chondrogenní tumory
V kapitole chrupavčitých nádorů došlo k několika ne příliš zásadním změnám. Nově uvedenou jednotkou je velmi vzácný benigní tumor, osteochondromyxom, charakterizovaný produkcí chrupavčité a osteoidní matrix, místy nápadně myxoidně změněné. Vyskytuje se v kostech splanchnokrania a v tibii a bývá asociován s Carneyho komplexem. Někdy může růst lokálně agresivně.
Částečně analogicky s nomenklaturou lipomatózních tumorů je nově pro chondrosarkom grade 1 zaveden synonymický termín atypický chrupavčitý tumor s nově přiděleným MKN-O kódem 9222/1. Dobře diferencované chondrogenní neoplázie rostou lokálně agresivně, ale metastazují jen velmi zřídka. Léčeny jsou tedy jen chirurgickou resekcí.
Osteogenní tumory
Fibrogenní tumory
V těchto kategoriích nebyla učiněna žádná zásadní změna. K benigním osteogenním tumorům byl nově zařazen osteom, který v předchozí klasifikaci chyběl. Za zmínku stojí konstatování, že ve všech dosud testovaných konvenčních osteosarkomech nebyla detekována IDH1 nebo IDH2 mutace, která je naopak běžná u chondrosarkomů. Tento poznatek by mohl napomoci k jednoznačnému odlišení některých obtížně diagnostikovatelných případů chondrogenních osteosarkomů.
Fibrohistiocytární tumory
Tato kategorie byla redukována o jednotku tzv. MFH kostí, který je nyní přejmenován na high-grade nediferencovaný pleomorfní sarkom a v sekci kostních tumorů tvoří samostatnou skupinu.
Ewingův sarkom
Patří mezi maligní nádory s neuroektodermální diferenciací a je charakterizován recipročními balancovanými translokacemi, které ve většině případů zahrnují EWSR1 gen na chromozomu 22 s některým ze členů ETS rodiny genů, nejčastěji genem FLI1. Oproti očekávání bylo zjištěno, že přítomnost fúzního genu EWSR1-ETS typu nemá žádný prognostický význam.
Hematopoetické neoplázie
V této kategorii jsou samostatně uvedeny poměrně časté léze, plazmocelulární myelom a solitární plazmocytom kostí. Další primární ne-hodgkinské lymfomy kostí jsou shrnuty do jedné kapitoly, jako vzácně se vyskytující primární kostní neoplázie, z nichž relativně nejčastější je difúzní velkobuněčný B lymfom.
Na osteoklasty bohaté tumory
Nově je uvedena diagnóza obrovskobuněčného tumoru malých kostí, s alternativním názvem obrovskobuněčný reparativní granulom. Tato afekce postihuje krátké kosti rukou a nohou a typicky se vyskytuje u adolescentů jako bolestivá léze doprovázená zduřením příslušné oblasti. Histologický obraz je podobný obrovskobuněčnému kostnímu tumoru.
Notochordální tumory
V této kategorii je poprvé představen pojem benigní notochordální nádor, který koresponduje s již vžitými názvy identické afekce (notochordální zbytky, notochordální hamartom, v případě sfenookcipitální lokalizace je užíván termín ecchordosis physaliphora). Většinou se jedná o lézi milimetrových rozměrů, histologicky tvořenou fyzaliforními buňkami s drobnými jádry, bez mitóz, nekróz, myxoidního prosáknutí a bez fibrózních pruhů. Je diskutována možnost transformace benigního notochordálního tumoru do chordomu, která pokud existuje, zahrnuje zcela raritní případy (11). Dle recentních studií chybí v chordomech IDH1 a IDH2 mutace, což může ve sporných případech sloužit pro odlišení chordomu od chondrosarkomu.
Vaskulární tumory
Byly rozšířeny o diagnózy epiteloidního hemangiomu a epiteloidního hemangioendoteliomu, které se mohou primárně v kostech také vyskytovat. První jmenovaná jednotka je řazena mezi tumory s intermediárním biologickým potenciálem, s rizikem lokální recidivy i možností vzdáleného metastazování. Epiteloidní hemangioendoteliom patří mezi maligní nádory s obtížně predikovatelnou prognózou, kterou nelze stanovit na základě histopatologického vyšetření.
Myogenní, lipogenní a epiteliální tumory
Zůstaly beze změn.
Tumory s nedefinovaným neoplastickým původem
U přibližně 70 % primárních aneurysmatických kostních cyst lze detekovat přestavbu USP6 genu na chromozomu 17p13, což potvrdilo dřívější domněnku, že se ve většině případech primárních aneurysmatických kostních cyst jedná o neoplastický proces, nově tedy s přiřazeným morfologickým kódem MKN-O 9260/0. Vzhledem k nezanedbatelné tendenci k lokálním recidivám je řazena mezi intermediární, lokálně agresivní tumory.
High-grade nediferencovaný pleomorfní sarkom
Vyčleněním z tzv. fibrohistiocytárních tumorů vytvořil samostatnou kategorii. Zároveň byl tumor přejmenován. Termín MFH už by neměl být používán.
Nádorové syndromy
Kromě již dříve uvedených nádorových syndromů se mezenchymální tumory častěji vyskytují i u pacientů s Liovým-Fraumeniho syndromem, neurofibromatózou typu 1 a cherubismem.
ZÁVĚR
Nová WHO klasifikace tumorů měkkých tkání a kostí plynule navazuje na předchozí 3. vydání. Mezi nejdůležitější uvedené změny patří úplné odstranění termínu maligního fibrózního histiocytomu, dříve považovaného za samostatnou jednotku, a řazeného mezi tzv. „fibrohistiocytární“ tumory. Maligní mezenchymální tumory, u kterých nelze pomocí současných metod určit linii diferenciace, nyní tvoří samostatnou kategorii nediferencovaných / neklasifikovaných sarkomů a jsou dále subklasifikovány na základě morfologie utvářejících buněk. Nově jsou součástí 4. vydání také tumory z nervových obalů a gastrointestinální stromální tumory. Na základě detailnějšího poznání histogeneze došlo k přeuspořádání některých nádorů do jiných kategorií. V sekci měkkotkáňových i kostních tumorů bylo zařazeno několik nových jednotek. Výrazný nárůst množství genetických dat podtrhuje význam molekulárně genetických a cytogenetických metod, díky kterým se podařilo řadu jednotek správně zařadit do patřičné skupiny tumorů, některé diagnózy byly odhaleny jako neopodstatněné a z nové klasifikace odstraněny nebo sloučeny s jinými jednotkami. Diagnostika mezenchymálních lézí je tak reprodukovatelnější, což se odráží především v úspěšnosti léčebných protokolů. Lze očekávat, že s pokračujícími úspěchy molekulárních a genetických studií budou definovány další neoplastické jednotky a ty stávající budou lépe subklasifikovány.
Adresa pro korespondenci:
MUDr. Karel Veselý, Ph.D.
I. PAÚ LF MU a FN u sv. Anny v Brně
Pekařská 53, 656 91 Brno
tel.: 543 183 220
fax: 543 183 217
e-mail: karel.vesely@fnusa.cz
Zdroje
1. Fletcher CDM, Unni KK, Mertens F, et al. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon 2002.
2. Fletcher CDM. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology 2006; 48 : 3-12.
3. Fletcher CDM, Bridge JA, Hogendoorn PCW, et al. WHO classification of tumours of soft tissue and bone. 4th Ed, IARC Press: Lyon, 2013.
4. Fletcher CDM. The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology 2014; 64(1): 2-11.
5. Deyrup ET, Weiss SW. Grading of soft tissue sarcomas: the challenge of providing precise information in an imprecise world. Histopathology 2006; 48 : 42-50.
6. Chibon F, Lagarde P, Salas S, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med 2010; 16 : 781-787.
7. Lagarde P, Perot G, Kauffmann A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res 2012; 18 : 826-838.
8. Mills AM, Beck AH, Montgomery KD, et al. Expression of subtype-specific group 1 leiomyosarcoma markers in a wide variety of sarcomas by gene expression analysis and immunohistochemistry. Am J Surg Pathol 2011; 35 : 583-589.
9. Bosman FT, Carneiro F, Hruban HR, et al. WHO classification of tumours of the digestive system. IARC: Lyon 2010; 74-76.
10. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23(2): 70-83.
11. Yamaguchi T, Watanabe-Ishiiwa H, Suzuki S, Igarashi Y, Ueda Y. Incipient chordoma: a report of two cases of early stage chordoma arising from benign notochordal cell tumors. Mod Pathol 2005; 18 : 1005-1010.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2014 Číslo 2
Najčítanejšie v tomto čísle
- WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition
- Kde končí a začíná diagnóza Ewingova sarkomu - popis dvou neobvyklých kostních nádorů s translokací t(20;22)(EWSR1-NFATc2)
- Současný staging zhoubných nádorů děložního těla a jeho význam pro klinickou praxi
- Epidermolytická hyperkeratóza vulvy asociovaná s bazocelulárním karcinomem u pacientky s vaginálním condyloma acuminatum a vaginální intraepiteliální neoplazií infikovanými HPV typu 42