
Molekulární patologie plicních karcinomů
Molecular pathology of pulmonary carcinomas
The group of non–small cell lung carcinomas includes tumors that are variable at the clinical, histopathological and molecular levels. Advances in the understanding of molecular pathology of lung adenocarcinomas in particular has led to changes in their histopathological classification and treatment. Patients diagnosed with lung adenocarcinoma harboring specific mutations benefit from the administration of specific targeted therapy. Therefore, pathologists closely involved in the diagnostics of lung tumors significantly contribute to the diagnostic-therapeutical algorithm. Analysis of EGFR gene mutations in lung adenocarcinomas is already routinely performed and the presence of activating mutations in EGFR is the main indication for the administration of tyrosinkinase inhibitors. Besides EGFR mutations, EML4–ALK rearrangement is also being analysed and there is potential in analysing BRAF mutations as well. The aim of this review is to summarize the role of the most relevant molecules that also serve as the therapeutic target for practicing pathologists.
Keywords:
NSCLC – lung adenocarcinoma – EGFR – ALK – BRAF – KRAS – RET – MET – erlotinib – gefitinib – crizotinib
Autoři:
Zdeněk Rohan 1,2; Milada Matějčková 1; Radoslav Matěj 1,2
Působiště autorů:
Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
1; Ústav patologie, Univerzita Karlova v Praze, 3. lékařská fakulta
2
Vyšlo v časopise:
Čes.-slov. Patol., 50, 2014, No. 2, p. 71-75
Kategorie:
Přehledový článek
Souhrn
Nemalobuněčné plicní karcinomy jsou typické variabilitou nejen ve svém klinickém průběhu a histopatologickém obraze, ale i v molekulárně-genetických charakteristikách. Zejména u primárních plicních adenokarcinomů vedly nové poznatky na poli molekulární genetiky k zásadní změně v přístupu k diagnostice a léčbě. Identifikace charakteristických genových aberací umožnila u části těchto nádorů nasazení tzv. cílené (biologické) léčby a tím zlepšení prognózy pacientů. S výše uvedeným zároveň vzrostla role molekulární patologie a zejména diagnostikujících patologů, kteří se správnou diagnózou a účelným nakládáním s nádorovou tkání aktivně podílí na diagnosticko-terapeutickém procesu. V současné době se již rutinně detekují mutace v genu pro receptor epidermálního růstového faktoru – EGFR, jejichž přítomnost je racionální indikací k nasazení cílené léčby inhibitory tyrosinkináz. Mimo EGFR se do rutinní diagnostické praxe zavedla detekce přestavby EML4–ALK, dalším potenciálním diagnostickým cílem jsou mutace genu kódujícího protein b-raf – BRAF. Sdělení shrnuje základní poznatky o diagnosticky a prakticky relevantních molekulách, které se podílejí na patogenezi vzniku plicních karcinomů a zároveň jsou cílem moderní, cílené terapie.
Klíčová slova:
NSCLC – plicní adenokarcinom – EGFR – ALK – BRAF – KRAS – RET – MET – erlotinib – gefitinib – crizotinib
Z molekulárně-genetického hlediska jsou pro prakticky všechny solidní nádorové afekce typické změny v expresi a aktivitě genů a proteinů zapojených v regulaci buněčného růstu, diferenciace, buněčné smrti, motility, responsivity k růstovým faktorům a v komunikaci s okolními buňkami a extracelulární matrix. Často se jedná o tzv. onkogeny a tumor supresorové geny, z nichž je část mutována u řady nádorů a jsou tak využívány jako markery nádorové transformace buňky (např. proteiny p53 a Rb1). Svou nespecificitou a komplexitou interakcí v procesu nádorové transformace ale omezují praktické využití cílené molekulárně-genetické diagnostiky i následné cílené terapie. I proto je cílem současného snažení detailněji poznat molekulárně-genetické změny specifických onkogenů a tumor supresorových genů charakteristických pro určitou skupinu či typ nádoru a tím zpřesnit nejen diagnostiku, ale i potenciální léčbu. V případě plicních nádorů se toto podařilo u části primárních plicních adenokarcinomů, u kterých byly popsány charakteristické mutace v EGFR, BRAF a přestavby zahrnující ALK, proti kterým je v současné době dostupná cílená terapie, ať už v rutinní praxi, či ve fázi klinických studií.
NEMALOBUNĚČNÉ PLICNÍ KARCINOMY (NSCLC)
EGFR
Receptor pro epidermální růstový faktor (EGFR – epidermal growth factor receptor) je kódován na krátkém raménku 7. chromozomu (7p11.2) a patří do HER/erbB rodiny transmembránových receptorů s cytoplazmatickou tyrosinkinázovou aktivitou, která zahrnuje HER1 (EGFR/erbB1), HER2 (neu/erbB2), HER3 (erbB3) a HER4 (erbB4). Podstatou aktivace EGFR je vazba ligandu (např. EGF, TGF-α) na extracelulární doménu receptoru, což vede ke změně jeho konformace a vzniku homodimerů (EGFR–EGFR) či heterodimerů (např. EGFR–neu). Tato konformační změna je zásadní pro následnou aktivaci cytoplazmatické tyrosinkinázové domény, která pak dále interaguje s dalšími (tzv. adaptorovými) molekulami s návazností řady signálních kaskád. V současné době jsou popsány tři základní signální dráhy EGFR: 1) RAS-RAF, 2) PI3K-AKT-mTOR a 3) JAK-STAT (Obr. 1). Aktivace EGFR má za následek řadu změn v genové a proteinové expresi buňky, což vede, z hlediska nádorové transformace, zejména k podpoře proliferace, motility, angiogeneze, invazivního růstu, odolnosti vůči buněčné smrti a vzniku metastatického fenotypu buněk (1).

Způsob, jakým může být jinak velmi precizně regulovaná funkce EGFR narušena, je založen na zvýšené (konstitutivní) aktivitě tyrosinkinázové domény EGFR. Tento patogenní mechanismus je rovněž podpořen faktem, že podstatná většina doposud známých proonkogenních mutací EGFR je právě v oblasti tyrosinkinázové domény. Na zvýšené aktivitě EGFR se však dále může podílet i amplifikace genu na úrovni DNA. A je to právě hyperaktivní tyrosinkinázová doména, proti které je namířena cílená terapie plicních karcinomů s prokázanou mutací EGFR – tzv. inhibitory tyrosinkináz (TKI – tyrosin kinase inhibitors), v současnosti zahrnující preparáty gefitinib a erlotinib.
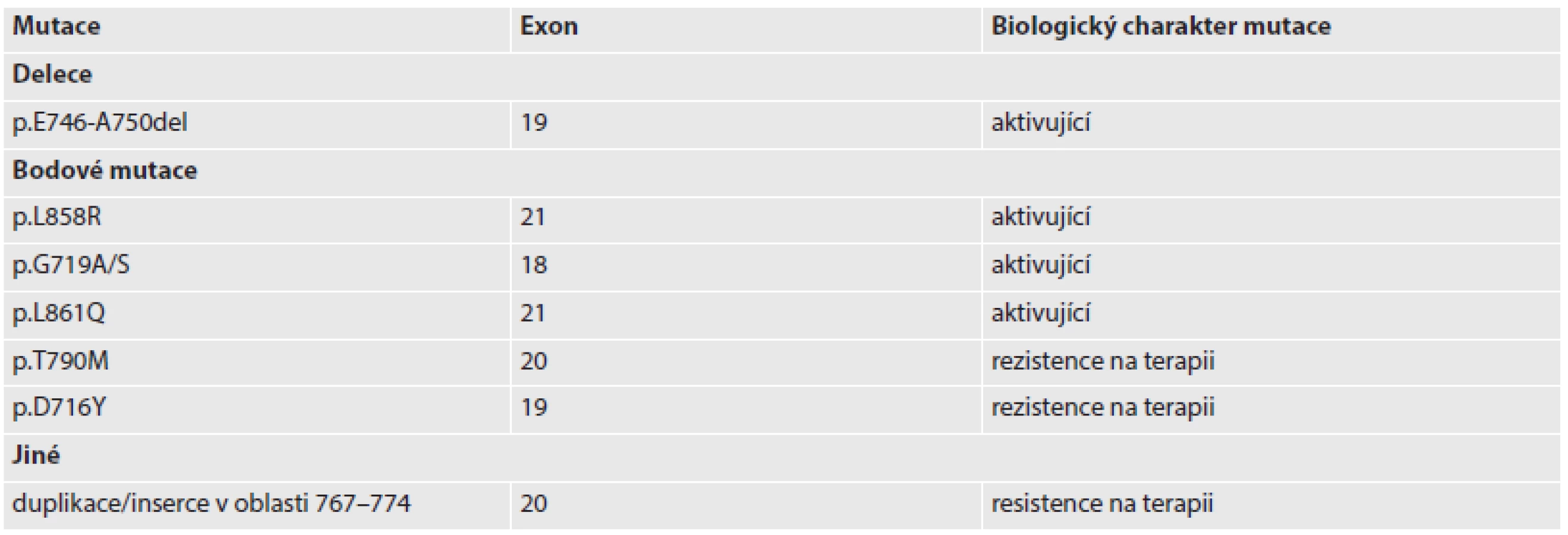
Přestože jsou mutace v EGFR poměrně častým jevem nejenom u plicních karcinomů, mutace lokalizované v exonech 18 – 21, které kódují tyrosinkinázovou doménu EGFR, jsou pro NSCLC specifické a nebyly detekovány v karcinomech tlustého střeva, pankreatu, prostaty, močového měchýře, prsu a ani v neuroendokrinních karcinomech (2). Podle databáze COSMIC (Catalogue of Somatic Mutations in Cancer – http://cancer.sanger.ac.uk) jsou v době sepisování tohoto sdělení mutace v EGFR u primárního plicního adenokarcinomu přítomny až u 35 % z celkového počtu přibližně 26 000 analyzovaných vzorků, což je druhé místo hned po mutacích v genu TP53 (38 % z přibližně 1 900 vzorků). Na třetím místě jsou pak mutace v onkogenu KRAS (20 % z přibližně 11 000 vzorků). Tato čísla jsou však hrubě orientační a zejména v případě mutací EGFR je frekvence mutovaných alel významně závislá na studované populaci – v asijské populaci jsou přítomny u 30 – 50 %, u populace západní je jejich udávaná frekvence 10 – 20 % (3). Mezi nejčastější mutace EGFR patří delece v exonu 19 (nejčastěji p.E746_A750del) a bodové (tzv. missense) mutace vedoucí k náhradě jednotlivých aminokyselin v sekvenci proteinu (zejména mutace p.L858R v exonu 21). Mimo bodové mutace, které vedou v případě EGFR k záměně aminokyselin, jsou popisovány i amplifikace EGFR.
Z prediktivního hlediska jsou důležité mutace EGFR v exonu 20, které vedou k resistenci nádoru k léčbě inhibitory tyrosinkináz. Primární rezistence nádorů k terapii je typicky podmíněna duplikacemi nebo inzercemi v exonu 20. Pro sekundární rezistenci, tzn. rezistenci navozenou klonální selekcí nádorových buněk s danou mutací, je typická mutace p.T790M, která vede ke sterickému narušení vazebného místa pro molekuly inhibitorů tyrosinkináz (4,5). Zajímavé je, že tyto rezistentní mutace typicky vznikají na stejné alele genu pro EGFR, která nese mutaci aktivující (tzv. cis mutace). Rezistence vůči inhibitorům tyrosinkináz však nemusí být způsobena pouze mutacemi v sekvenci EGFR. Bylo zjištěno, že amplifikace či zvýšená exprese tyrosinkinázy MET (viz níže), vede k posílení PI3K-AKT a RAS-RAF signálních kaskád a tím „obejde“ vliv inhibitorů EGFR (obr. 1) (6).
Primární plicní karcinomy nesoucí mutaci EGFR jsou charakteristické svým výskytem u osob ženského pohlaví bez anamnézy chronického nikotinismu, vyšší výskyt je popisován u asijské populace (3). Mutace EGFR byly doposud prakticky výhradně popisovány u primárních plicních adenokarcinomů, spíše ojediněle pak byly popsány i v adenoskvamózním karcinomu a zcela výjimečně i u primárního plicního skvamocelulárního karcinomu, přičemž nebyly popsány u mucinózních, „salivary-gland type“ a neuroendokrinních karcinomů plic. (7,8) Typický adenokarcinom s mutací v EGFR je morfologicky charakterizován lepidickým typem růstu spolu s acinární či papilární složkou, bez nekróz a bez výraznějších cytologických atypií. V imunohistochemickém vyšetření jsou tyto nádory TTF-1 a napsin A pozitivní a p63/p40 negativní (3,7,9).
Molekulárně-genetická diagnostika mutací se v současné době opírá zejména o sekvenaci genu EGFR. Tato metoda je však optimální pro záchyt mutací, je-li ve vzorku více než 50 % nádorových buněk. Pro rutinní diagnostickou praxi však existují metody a postupy, kterými lze senzitivitu zvýšit (např. makro-/mikrodisekce, real-time PCR, sekvenování nové generace atd.) a jsou tedy výhodnější a výtěžnější. Metody využívající in situ hybridizace (FISH) nejsou pro detekci mutací EGFR vhodné, i když je známo, že EGFR může být v nádorech amplifikován. V provedených studiích neměl počet kopií EGFR prediktivní ani prognostický význam. Většímu zájmu se těší imunohistochemická metoda detekce pomocí monoklonálních protilátek namířených proti specifickým nejčastějším mutacím molekul EGFR, nicméně výsledky doposud provedených studií nejsou jednoznačné (10-12). Imunohistochemické vyšetření zatím tedy nelze považovat za adekvátní a plnohodnotnou náhradu molekulárně-genetického vyšetření genu EGFR.
ALK
Přestavby zahrnující ALK (anaplastic lymphoma kinase; lokus 2p23) byly poprvé popsány u anaplastického velkobuněčného lymfomu (ALCL, přestavba NPM–ALK). Dále byly nalezeny například u inflamatorního myofibroblastického tumoru (13,14), některých karcinomů ledviny (15) a u přibližně 5 % NSCLC (16). Nejčastěji popisovaná přestavba u NSCLC je delece a následná inverze na krátkém raménku 2. chromozomu – inv(2)(p21p23), která vede ke vzniku fúzního produktu EML4–ALK (EML – echinoderm microtubule associated protein-like 4) (17). Možností přestavby je hned několik, přičemž se liší délka EML4 části fúzního genu, délka ALK zůstává stejná (16). Přestavby zahrnující ALK se až na jednotlivé výjimky ve většině případů navzájem vylučují s mutacemi EGFR. (18)
Klinicky se NSCLC s přestavbou EML4–ALK často diagnostikují v pokročilém stadiu a spíše u nekuřáků nebo lehkých kuřáků ve věku 40–60 let. Histopatologicky se jedná o hůře diferencované adenokarcinomy charakterizované solidním a/nebo acinárním růstem, nekrózami, často s nápadnou příměsí lymfocytů i přítomností buněk vzhledu pečetního prstenu. Rovněž může být přítomna skvamocelulární komponenta, či se může jednat i o adenoskvamózní karcinom, přestavba EML4–ALK byla popsána i u mukoepidermoidního karcinomu. Imunohistochemický profil odpovídá nádorům odvozeným z tzv. terminální rezervní jednotky – jsou TTF-1 a napsin A pozitivní a p63/p40 a CK5/6 negativní (3,17-20). Zajímavé je, že v jedné studii byla u části buněk s morfologií buněk typu pečetního prstenu pozorována koexprese TTF-1 a p63 (21).
Diagnostika přestavby EML4–ALK na molekulární úrovni se opírá zejména o metody in situ hybridizace (FISH), rovněž se do praxe začíná zavádět nepřímá imunohistochemie využívající monoklonální protilátky se zvýšenou senzitivitou i specificitou pro ALK přestavby (22-24).
I přes relativně nízký výskyt jsou NSCLC s přestavbou EML4–ALK v popředí zájmu, a to vzhledem k dostupnosti účinného inhibitoru ALK, crizotinibu, který v klinických studiích prodloužil přežívání pacientů s touto aberací (25).
BRAF
Protein BRAF (v-raf murine sarcoma viral oncogene homolog B) je serin/threoninová proteinová kináza, která je zapojena v signální kaskádě RAS-RAF. Aktivující mutace BRAF postihující kinázovou doménu (ve většině případů mutace p.V600E), jsou časté u melanomu (26) a byly rovněž popsány u dalších nádorů včetně kolorektálního karcinomu (26) a papilárního karcinomu štítné žlázy (27), u NSCLC tvoří minoritní podíl – přibližně 3 % (28). Vzhledem k dostupnosti již registrovaného inhibitoru mutovaného BRAF (p.V600E), vemurafenibu, který se užívá v léčbě pokročilých stadií maligního melanomu, je analýza BRAF statutu u pacientů s plicním adenokarcinomem dalším možným krokem.
KRAS
Protein KRAS (Kirsten rat sarcoma viral oncogene homolog) patří do rodiny RAS onkogenů (spolu s NRAS a HRAS), jejichž aktivita je regulována vazbou a štěpením GTP (guanosin trifosfát). Zjednodušeně řečeno, aktivovaný KRAS má na sobě navázaný GTP, který je následně hydrolyzován na GDP a tím se KRAS inaktivuje. Podstatou onkogenní aktivity KRAS jsou mutace inhibující hydrolýzu GTP, což vede ke zvýšené aktivitě KRAS. Protein KRAS je zapojen jako jeden z hlavních regulátorů signální dráhy RAS-RAF a je také zapojen do signalizace PI3K-AKT-mTOR (29).
Mutace KRAS jsou popisovány u celé řady nádorů zahrnující kolorektální karcinomy, karcinomy plic, prsu, močového měchýře, žaludku a pankreatu, vrozené mutace KRAS jsou typické pro u kraniofaciokutánní syndrom 2 (OMIM# 190070; www.omim.org). Přibližně 20 % primárních plicních adenokarcinomů nese mutaci KRAS, u skvamocelulárních karcinomů či u malobuněčného karcinomu jsou mutace KRAS popisovány zcela výjimečně. Plicní adenokarcinomy s mutovaným KRAS se vyskytují spíše u mužů kuřáků a je pro ně typická kratší doba přežití (30,31). Typicky se jedná o solidně rostoucí adenokarcinomy s nekrózami, buněčnými atypiemi a přítomností tumor-infiltrujících leukocytů. Pozitivita TTF-1 byla pozorována u většiny adenokarcinomů s mutovaným KRAS non-mucinózního typu; u mucinózních adenokarcinomů bylo TTF-1 většinou negativní (9).
Přítomnost mutace KRAS u plicních karcinomů lze považovat za prognostický marker; její prediktivní hodnota je značně omezená (30,31). Současné poznatky navíc ukazují, že mutace v EGFR a KRAS se navzájem vylučují a tudíž lze předpokládat, že je-li přítomna mutace v EGFR, je riziko primární rezistence proti EGFR inhibitorům založené na konstitutivní aktivitě mutovaného KRAS zanedbatelné (32).
Další potenciální markery
Přibližně u 1 – 7 % pacientů s primárním plicním adenokarcinomem lze nalézt amplifikaci onkogenu MET, což je receptor s tyrosinkinázovou aktivitou (známý také jako HGFR – hepatocyte growth factor receptor) spřažený se signálními drahami RAS-RAF a PI3K-AKT-mTOR (obr. 1) (6). Jeho role jako onkogenního aktivátoru výše uvedených drah je analogická EGFR, navíc je zvýšená aktivita MET podkladem až 20 % případů sekundární rezistence k léčbě inhibitory tyrosinkinázové aktivity EGFR (6).
Další genetickou alterací identifikovanou u plicních adenokarcinomů je přestavba KIF5B–RET (KIF5B – kinesin family member 5B) (33). Onkogenem je zde RET, receptor s tyrosinkinázovou aktivitou, jehož aktivující mutace jsou známy u syndromů MEN 2A a MEN 2B, ztráta funkce RET je popisována v rámci patogeneze Hirschsprungovy choroby. (34)
Zajímavá je nově popsaná přestavba zahrnující onkogen ROS1, který je svou aktivitou podobný ALK. Podoba však nezůstává pouze u aktivity, ale stejně jako pro ALK bylo i pro ROS1 nalezeno několik fúzních partnerů a také morfologie „ALK-“ a „ROS1-adenokarcinomů“ si jsou velmi podobné. (35) Tato podobnost s ALK může být potenciálním benefitem při užití inhibitoru ALK crizotinibu.
SKVAMOCELULÁRNÍ PLICNÍ KARCINOM
V případě dalšího významného člena skupiny tzv. nemalobuněčných plicních karcinomů, karcinomu skvamocelulárního, je již poznání molekulárního pozadí onkogeneze omezenější. Skvamocelulární karcinom plic je etiologicky nejčastěji spojován s působením karcinogenů vzniklých spalováním tabáku a cigaretového papíru často velmi pochybných kvalit a složení, a proto lze očekávat, že genové a proteinové alterace budou u těchto nádorů velmi variabilní, mnohočetné a poměrně nespecifické. I proto jsou výsledky molekulárně-genetických a genomických studií a jejich následné převedení do konkrétních diagnostických a léčebných postupů stále problematické. Přesto lze předpokládat existenci přítomnosti společných molekulárních jmenovatelů typických pro metaplastický, dysplastický následně i invazivní fenotyp těchto lézí bronchiálního epitelu (36). Nejčastěji se v tomto kontextu popisují amplifikace genu pro FGFR1 (fibroblast growth factor receptor 1) a mutace v FGFR2, DDR2 (discoidin domain receptor tyrosine kinase 2) a PIK3CA (phosphatidylinositol-bisphosphate 3-kinase). V současnosti probíhají klinické zkoušky s inhibitory FGFR1 a DDR2, u kterých byla u pacientů se skvamocelulárním karcinomem popsána velmi nadějná terapeutická odpověď, nicméně tyto výsledky je zatím třeba hodnotit s nadhledem a vyčkat dalších prospektivních studií a metaanalýz (36,37).
MALOBUNĚČNÝ PLICNÍ KARCINOM
Malobuněčný plicní karcinom stojí zcela stranou výše diskutovaných nemalobuněčných karcinomů plic. Jeho agresivní chování, potenciální systémové postižení již v době diagnózy a tím velmi nepříznivá prognóza si vyžadují zcela odlišných terapeutických postupů. Jedinou léčbou jsou chemoterapie s radioterapií, nicméně i po nich nádor rychle recidivuje a procento 5-letého přežívání přes veškerý pokrok v léčebných modalitách se limitně blíží nule. Na poli molekulární genetiky je jejich charakterizace poměrně omezená a zatím jsou stabilně popisovány mutace v genech TP53, RB1 a PTEN.
ZÁVĚR A SOUČASNÉ STANDARDY VYŠETŘOVÁNÍ
Diagnostika plicních karcinomů prošla během posledních několika let řadou změn ať už na poli klasifikace plicních adenokarcinomů či charakteristiky cílů specifické terapie. Změna histopatologické klasifikace a diagnostického algoritmu při vyšetřování plicních adenokarcinomů je shrnuta v mezinárodních konsensech (32,38) a v Doporučeném postupu pro histologické vyšetření karcinomu plic vydaném v roce 2013 Společností českých patologů (dostupný ke stažení na www.patologie.info/standardy.php).
V současnosti je doporučováno u všech nádorů určených jako primární plicní adenokarcinom provést analýzu mutací EGFR a na vyžádání ošetřujícího pneumoonkologa přestavby EML4–ALK. Analýza mutací KRAS u plicních adenokarcinomů není v současné době z důvodů uvedených výše v textu indikováno. Jako optimální metody pro diagnostiku mutací EGFR se jeví izolace DNA z nádorové tkáně a následná analýza potenciálních patogenních variací genu EGFR pomocí metody real-time PCR (obr. 2A), případně reverzní hybridizace produktů PCR na tzv. „stripu“ (obr. 2B) nebo sekvenace genu EGFR na přístroji druhé generace sekvenátorů, tzv. NGS (NGS – Next-Generation Sequencing). Molekulárně-genetickou analýzu lze provést jak z bronchoskopického vzorku fixovaném ve formolu a zalitém do parafinu tak i z cytologického nátěru. Současné metody umožňují detekovat 1 % mutované DNA na pozadí nemutované, proto lze analyzovat i vzorky tkáně, ve kterých je zastoupení nádorových buněk malé. Prakticky je však žádoucí a v doporučených postupech vyžadované zastoupení alespoň 10 % nádorové tkáně. Přítomnost přestavby EML4–ALK lze diagnostikovat buď pomocí metod využívajících in situ hybridizace (FISH, CISH, SISH), nebo imunohistochemicky pomocí monoklonální protilátky namířené proti EML4–ALK.


Právě optimalizace postupů a multidisciplinární přístup k diagnostice NSCLC spočívající v úzké spolupráci pulmoonkolooga, histopatologa a molekulárního biologa vede ke zvýšení efektivity diagnostických postupů. Bez harmonizace a úzké spolupráce nelze dosáhnout optimálních výsledku, ba co více, je možné dojít k výsledkům zavádějícím a v konečném důsledku poškozujícím pacienta.
PODĚKOVÁNÍ
Práce byla podpořena grantem NT14406-3/2013 IGA MZd ČR a výzkumným projektem PRVOUK - Onkologie P27, uděleným Univerzitou Karlovou v Praze.
Adresa pro korespondenci:
Doc. MUDr. Radoslav Matěj, Ph.D.
Oddělení patologie a molekulární medicíny
Thomayerova nemocnice
Vídeňská 800, 14059 Praha 4 – Krč
tel.: 261 083 741
e-mail: radoslav.matej@ftn.cz
Zdroje
1. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 2013; 79C: 34-74.
2. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97(5): 339-346.
3. Yousem SA. Role of molecular studies in the diagnosis of lung adenocarcinoma. Mod Pathol 2012; 25 Suppl 1S11-17.
4. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2(3): e73.
5. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352(8): 786-792.
6. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104(52): 20932-20937.
7. Zakowski MF, Hussain S, Pao W, et al. Morphologic features of adenocarcinoma of the lung predictive of response to the epidermal growth factor receptor kinase inhibitors erlotinib and gefitinib. Arch Pathol Lab Med 2009; 133(3): 470-477.
8. Sartori G, Cavazza A, Sgambato A, et al. EGFR and K-ras mutations along the spectrum of pulmonary epithelial tumors of the lung and elaboration of a combined clinicopathologic and molecular scoring system to predict clinical responsiveness to EGFR inhibitors. Am J Clin Pathol 2009; 131(4): 478-489.
9. Rekhtman N, Ang DC, Riely GJ, Ladanyi M and Moreira AL. KRAS mutations are associated with solid growth pattern and tumor-infiltrating leukocytes in lung adenocarcinoma. Mod Pathol 2013; 26(10): 1307-1319.
10. Allo G, Bandarchi B, Yanagawa N, et al. Epidermal growth factor receptor mutation-specific immunohistochemical antibodies in lung adenocarcinoma. Histopathology 2013; 20. doi: 10.1111/his.12331. [Epub ahead of print]
11. Isaksson S, Bendahl PO, Salomonsson A, et al. Detecting EGFR alterations in clinical specimens-pitfalls and necessities. Virchows Arch 2013; 463(6): 755-764.
12. Wen YH, Brogi E, Hasanovic A, et al. Immunohistochemical staining with EGFR mutation-specific antibodies: high specificity as a diagnostic marker for lung adenocarcinoma. Mod Pathol 2013; 26(9): 1197-1203.
13. Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000; 157(2): 377-384.
14. Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T and Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999; 59(12): 2776-2780.
15. Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 2011; 24(3): 430-442.
16. Horn L and Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol 2009; 27(26): 4232-4235.
17. Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK Fusion Is Linked to Histological Characteristics in a Subset of Lung Cancers. Journal of Thoracic Oncology 2008; 3(1): 13-17.
18. Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res 2009; 15(16): 5216-5223.
19. Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol 2009; 22(4): 508-515.
20. Popat S, Gonzalez D, Min T, et al. ALK translocation is associated with ALK immunoreactivity and extensive signet-ring morphology in primary lung adenocarcinoma. Lung Cancer 2012; 75(3): 300-305.
21. Yoshida A, Tsuta K, Watanabe S, et al. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer 2011; 72(3): 309-315.
22. To KF, Tong JH, Yeung KS, et al. Detection of ALK rearrangement by immunohistochemistry in lung adenocarcinoma and the identification of a novel EML4-ALK variant. J Thorac Oncol 2013; 8(7): 883-891.
23. Han XH, Zhang NN, Ma L, et al. Immunohistochemistry reliably detects ALK rearrangements in patients with advanced non-small-cell lung cancer. Virchows Arch 2013; 463(4): 583-591.
24. Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res 2010; 16(5): 1561-1571.
25. Shaw AT, Kim D-W, Nakagawa K, et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. New England Journal of Medicine 2013; 368(25): 2385-2394.
26. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417(6892): 949-954.
27. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE and Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003; 63(7): 1454-1457.
28. Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011; 29(15): 2046-2051.
29. Chetty R and Govender D. Gene of the month: KRAS. J Clin Pathol 2013; 66(7): 548-550.
30. Sun JM, Hwang DW, Ahn JS, Ahn MJ and Park K. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One 2013; 8(5): e64816.
31. Riely GJ, Marks J and Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc 2009; 6(2): 201-205.
32. Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol 2013; 8(7): 823-859.
33. Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med 2012; 18(3): 375-377.
34. Pan ZW and Li JC. Advances in molecular genetics of Hirschsprung’s disease. Anat Rec (Hoboken) 2012; 295(10): 1628-1638.
35. Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012; 18(3): 378-381.
36. Drilon A, Rekhtman N, Ladanyi M and Paik P. Squamous-cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. Lancet Oncol 2012; 13(10): e418-426.
37. Kim HS, Mitsudomi T, Soo RA and Cho BC. Personalized therapy on the horizon for squamous cell carcinoma of the lung. Lung Cancer 2013; 80(3): 249-255.
38. Travis WD, Brambilla E, Noguchi M, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 2011; 6(2): 244-285.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2014 Číslo 2
Najčítanejšie v tomto čísle
- WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition
- Kde končí a začíná diagnóza Ewingova sarkomu - popis dvou neobvyklých kostních nádorů s translokací t(20;22)(EWSR1-NFATc2)
- Současný staging zhoubných nádorů děložního těla a jeho význam pro klinickou praxi
- Epidermolytická hyperkeratóza vulvy asociovaná s bazocelulárním karcinomem u pacientky s vaginálním condyloma acuminatum a vaginální intraepiteliální neoplazií infikovanými HPV typu 42