
Vzácná oční manifestace u podezření na Alportův syndrom
Rare Ocular Manifestation with Suspect Alport Syndrome
The authors mention a case report of a 13 year old girl with renal disease, who visited the outpatient Department of Pediatric Ophthalmology, University Hospital Brno with subjective complaints on decreased vision of both eyes.
Ophthalmologic examination showed physiological foveolar reflex on fundus and very discrete changes of the retinal pigment epithelium in macula, the fundus periphery was without pathology.
OCT images showed bilateral atrophy of central macula and changes at the level of the photoreceptors.
The authors describe a rare ocular manifestation of macular atrophy with suspect Alport syndrome, which strengthened the suspicion of this disease.
The authors also mention other possible ocular manifestations of Alport syndrome and compare the findings with the up to date international references.
Key words:
Alport syndrome, X heterozygot Alport syndrome, macular atrophy, lentikonus
Autoři:
I. Krejčířová 1; B. Varadyová 1; Z. Doležel 2; R. Autrata 1; J. Matušová 3; E. Gregorová 3
Působiště autorů:
Dětská oční klinika, Fakultní nemocnice, Brno, přednosta prof. MUDr. Rudolf Autrata, CSc., MBA
1; Pediatrická klinika, Fakultní nemocnice, Brno, přednosta prof. MUDr. Zdeněk Doležel, CSc.
2; Klinika nemocí očních a optometrie, Fakultní nemocnice U sv. Anny, Brno, přednosta doc. MUDr. Svatopluk Synek, CSc.
3
Vyšlo v časopise:
Čes. a slov. Oftal., 70, 2014, No. 3, p. 114-118
Kategorie:
Kazuistika
Souhrn
Autoři popisují kazuistiku 13leté dívky s renálním onemocněním, která se dostavila na ambulanci Dětské oční kliniky, FN Brno se subjektivní stížností oboustranného poklesu vizu.
Oftalmoskopicky na fundu patrný fyziologický foveolární reflex, v makule velmi diskrétní změny na úrovni RPE, ostatní nález do periferie bez patologie.
OCT obraz ukázal bilaterální atrofii makuly a změny na úrovni fotoreceptorů.
Autoři podrobně popisují vzácnou oční manifestaci makulární atrofie u Alportova syndromu. Upozorňují na souvislost této retinální afekce s AS a zdůrazňují důležitost správné diagnostiky přínosné zejména u heterozygotních žen, u kterých je variabilita X vázaného onemocnění velká a přítomnost této retinální či jiné anomálie může podpořit podezření na toto celkové onemocnění a napomoci k celkové diagnostice systémového onemocnění.
Autoři se dále věnují obecně možné oční manifestaci u pacientů s Alportovým syndromem a srovnávají nález s dostupnou světovou literaturou.
Klíčová slova:
Alportův syndrom, X vázaný Alportův syndrom, makulární atrofie, lentikonus
Úvod
Alportův syndrom (AS) představuje hereditární onemocnění, při kterém dochází k postižení funkce ledvinných glomerulů. Jejich dysfunkce spočívá ve ztenčení bazálních membrán následkem nesprávné syntézy α řetězců kolagenu IV. typu (3, 11, 12).
Příčinou AS je nejčastěji mutace genu pro α5 řetězec kolagenu IV (nejčastěji mutace genu COL4A5 na X chromozomu) a v 85 % případů jde tedy o X vázanou dědičnost. V méně obvyklých případech (10–15 %) je onemocnění způsobeno mutací obou alel genu pro α3 nebo α4 řetězec kolagenu IV, pak se jedná o autosomálně recesivní dědičnost (3, 11, 12, 13).
Incidence AS je přibližně 1 : 5000 živě narozených dětí (3, 8). U heterozygotních žen se vyskytuje, jak ukázaly nedávné studie, velká variabilita onemocnění, a to jak klinických, tak i diagnostických nálezů (12, 17). Zatímco 90 % postižených mužů postihuje chronická renální insuficience před 40. rokem života, u heterozygotních žen je to pouze 12 % z postižených (12).
U pacientů s klasickým AS je typické postižení renální funkce (projevuje se jako nefritický nebo nefrotický syndrom). Plně vyvinutý AS se také nazývá jako progresivní hereditární nefritida (3, 11). S AS bývají asociovány poruchy sluchu a právě také oční manifestace.
Mezi klinické příznaky podporující podezření na AS patří příznaky renální (mikrohematurie, proteinurie, hypertenze a renální insuficience), sluchové (oboustranná ztráta sluchu ve vysokých frekvencích) a příznaky oční (3, 11). Kromě bazální membrány ledvinných glomerulů patří mezi možné postižené tkáně také bazální membrána cortiho orgánu vnitřního ucha, bazální membrána pouzdra oční čočky, vnitřní limitující membrána sítnice (ILM) a bazální membrána retinální pigmentového epitelu (RPE), která tvoří Bruchovu membránu (8, 13).
Interní stanovení diagnózy spočívá ve fyzikálním vyšetření a anamnéze, detailní rodinné anamnéze a případné analýze moči u blízkých příbuzných, imunohistochemickém vyšetření bazální membrány v renální nebo kožní biopsii, vyšetření renální biopsie pomocí elekronového mikroskopu, molekulárně genetickém vyšetření genů COL4A3, COL4A4 a COL4A5. Pokud lékař objeví mikroskopickou hematurii, je třeba vždy pomýšlet na AS (3, 11).
Variabilita onemocnění u heterozygotních žen s X vázaným AS se projevuje jak v klinických, tak diagnostických nálezech. Poslední studie ukázaly, že histologické nálezy u ledvinné biopsie u heterozygotních žen mají široce variabilní vzhled s nespecifickými nálezy detekovanými pomocí světelné mikroskopie. Imunohistochemické vyšetření také vykazuje u heterozygotních žen variabilní nálezy. Senzoneurální postižení sluchu, zejména ve vysokých frekvencích, se u postižených mužů vyskytuje do věku 40 let u 90 %, oproti 10 % u heterozygotních žen (12, 19).
Léčba AS je pouze symptomatická a spočívá léčbě hypertenze a snížení proteinurie. Jediná možná terapie u terminální fáze renálního selhání při AS je v současné době hemodialýza nebo orgánová transplantace ledviny. V budoucnosti se uvažuje o léčení pomocí genové terapie (3, 11).
V roce 1927 popsal poprvé Alport společnou přítomnost hereditární nefritidy se senzoneurální nedoslýchavostí pozorovanou v několika generacích. V roce 1954 byla poprvé pozorována deformace čiré čočky u pacientů s renálním a sluchovým postižením. V roce 1961 Julien Marie popsala přítomnost předního lentikonu u pacientů s AS (8).
Mezi nejčastější oční manifestace u AS patří přední lentikonus a skvrnitá perimakulární nebo periferní retinopatie („fleck and dots retinopathy“). Jako vzácné oční abnormality byly popsány zadní lentikonus, zadní polymorfní dystrofie rohovky, makulární atrofie a makulární díra (2, 4, 6, 7, 8, 9, 13, 16, 19). Oční vyšetření a nález oční manifestace může napomoci diagnostice AS (18).
Kazuistika
Na konci roku 2012 se 13letá dívka dostavila na ambulanci Dětské oční kliniky se subjektivní stížností oboustranného poklesu vizu. Oční osobní anamnéza bezvýznamná. Dívka je v pravidelné dispenzární péči Pediatrické kliniky FN Brno od roku 2010 pro nefrotický syndrom, kdy za poslední 2 roky prodělala šestkrát relaps tohoto onemocnění. Nyní 2 roky na celkové kortikosteroidní léčbě Prednisonem p.o., v období remise na minimálních dávkách 5–10 mg obden.
V roce 2010 byla provedena biopsie ledvin se závěrem nefrotický syndrom s obrazem minimálních změn glomerulů, s nespecifickým nálezem pro AS. V období remise nefrotického syndromu dívka zcela bez nálezu mikroskopické hematurie a proteinurie. Ve fázi zhoršení vždy obraz typicky rozvinutého nefrotického syndromu. Před nálezem oční manifestace nebylo zatím genetické vyšetření pro AS z interního hlediska indikováno.
Z očního klinického vyšetření vizus OD 0,9 d.k.n; vizus OS 0,7 d.k.n, dle cykloplegické refrakce emetropie, nitrooční tlak v normě, postavení paral., hybnost neomezena, zornice izokorické, bez známek aferentního pupilárního defektu. Přední segment s biomikroskopicky zcela s fyziologickým nálezem, bez patologického nálezu na rohovce nebo čočce.
Na fundu papila fyziologického vzhledu, cévy fyziologického průběhu i plnění, makula se zachovalým fyziologickým foveolárním reflexem, patrné jen diskrétní změny na úrovni retinálního pigmentového epitelu. Fotografická dokumentace fundu pravého a levého oka pacientky je znázorněna na obrázku 1 a, b.

Vyšetření na Amslerově mřížce bilaterálně bez patologie.
Proveden statický perimetr, kde vlevo patrný drobný relativní centrální skotom. Dle VEP jen hraniční latence vlny P100 bez významné patologie
Dívka následně odeslána na vyšetření OCT. Vyšetření provedeno na Spectral OCT SLO OPKO. Zjištěna oboustranná makulární atrofie zejména centrální části vzhledem k věku (1,5) se změnami na úrovni RPE a fotoreceptorů na obr. 2 a v tabulce 1.


Dále doplněno přístrojové vyšetření barvocitu (HMC anomaloskop, Oculus), který byl oboustranně bez postižení. Multifokální elektroretinografie ukázala výrazné deprese potenciálů makulární oblasti bilaterálně s prodlouženou latencí vlny P, což potvrzuje atrofii v centrální krajině. Výsledek analýzy laserové skenovací polarimetrie (GDx) bilaterálně v mezích normy.
Obr. 2a, 2b zobrazuje OCT makulární oblasti pravého a levého oka (ODS): Foveální deprese zachována, patrné umbo, patrné ztenčení celé makulární oblasti sítnice vzhledem k věku, zejména v centrální části, patrná separace RPE a hranice zevních a vnitřních segmentů fotoreceptorů.


Dívka byla odesílána i na vyšetření fluorescenční angiografií, retinálním specialistou však nebylo toto vyšetření indikováno pro dostačující nález v OCT obraze.
I když klinický obraz renálního onemocnění ne zcela typicky odpovídá AS, s absencí mikroskopické hematurie v období remise nefrotického syndromu a bez typického nálezu při biopsii ledvin, nález oční makulární patologie přispěl k potvrzení podezření na X vázaný AS. Anamnesticky doplněna přítomnost možného renálního onemocnění u babičky a matky dívky. U dívky doplněno audiologické a ORL vyšetření se závěrem normacusis, dívka tedy bez postižení sluchu.
Fenotypický obraz, dle nedávných studií (12, 9) sledujících heterozygotní ženy s X vázaným AS, může odpovídat právě velké variabilitě AS, kdy ani absence mikrohematurie a nespecifický nález biopsie ledvin nevylučuje toto onemocnění (12, 19).
Oční nález t.č. uzavřen jako vzácná oční manifestace makulární atrofie u podezření na AS. Z interního hlediska následně indikována DNA analýza na přítomnost mutace COL4A genů pro suspektní X vázaný AS. Vzorek k DNA analýze byl standardně zaslán do laboratoře Oddělení lékařské genetiky FN Ostrava, která vyšetření AS zajišťuje. V současné době, tj. více než rok po odběru, nemáme výsledek DNA analýzy k dispozici.
Diskuse
Hlavní oční abnormality u nemocných s AS jsou přední lentikonus, a centrální a periferní skvrnitá retinopatie. Tyto dvě nejčastější oční manifestace se vyskytují přibližně u 50 % mužů a 20 % žen s X vázaným AS (8, 13). Přední lentikonus je způsobem vyklenutím čočky v důsledku ztenčeného pouzdra čočky (13).
Centrální retinopatie zahrnuje bělavě žlutavé perimakularní tečky a skvrny, které jsou často přítomny od počátku dospívání a jsou častější, pokud je přítomna renální dysfunkce, postižení sluchu a přední lentikonus (13). Centrální a periferní retinopatie se však mohou vyskytnout i samostatně bez přítomnosti předního lentikonu u nemocných s AS a většinou neovlivňují vidění a nevyžadují léčbu (13). Centrální retinopatie se pohybuje od několika roztroušených bodů a skvrnek v temporální části makuly až po (v nejtěžších případech) perimakularní mezikruží složené z hustě nakupených bodů od okrajů fovey až po vzdálené cévní arkády. Mohou pak vytvářet specifické pruhy, které odrážejí uspořádání vrstvy svazků nervových vláken (13). Někdy tečky a skvrny produkují abnormální tapetální reflex (13) a jejich perifoveálním uspořádáním tak mohou ovlivňovat vlastní reflex foveální a foveolární, v literatuře „lozenge or dull macular reflex“ (2, 13).
U postižených AS je retinopatie zasahující foveu a makulární atrofie popisována vzácně (13). U pacientů s AS byly popsány i vzácné případy ztenčení sítnice s nálezem podobným dystrofii čípků (14, 15) či bull´s eyes makulopatii nebo viteliformní makulopatii (6). Makulární díry spojené s AS jsou taktéž raritní, jsou obvykle větší než klasické idiopatické makulární díry a špatně reagují na chirurgickou léčbu (13, 9, 10).
Tabulka 2 dokumentuje autory popisující případy ztenčení sítnice u pacientů s AS.
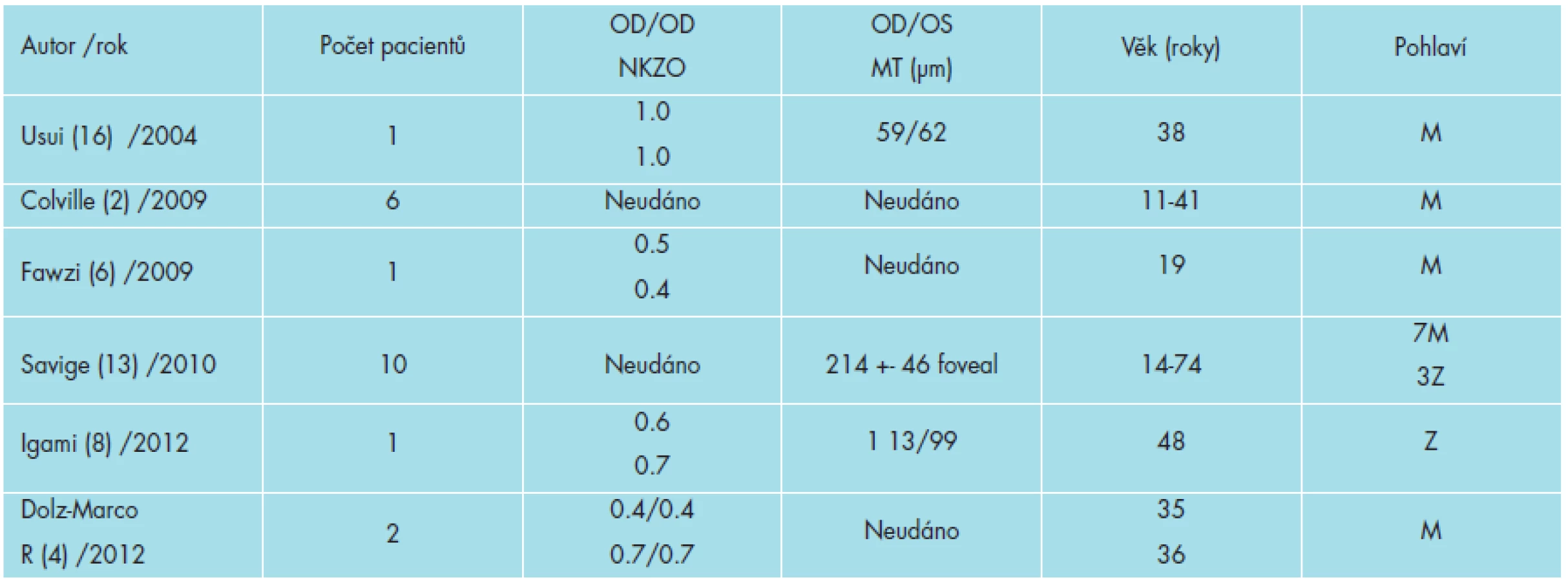
Přítomnost řetězců kolagenu IV typu byla imunohistochemickou analýzou prokázána v sítnici (13). Byly prokázány v ILM a v bazální membráně RPE Bruchovy membrány. U pacientů s AS bylo prokázáno ztenčení ILM /vrstvy nervových vláken a Bruchovy membrány (13). Perimakulární „fleck and dots“ – skvrnitá retinopatie odpovídá hyperreflexivitě ILM /vrstvě nervových vláken (13). Makulární díra a makulární atrofie má predilekci v temporální makulární oblasti (4, 8, 10, 13).
Předpokládá se, že retinální abnormality AS jsou výsledkem ztenčení vrstvy ILM /vrstvy nervových vláken a RPE bazální membrány Bruchovy membrány. Ztenčení těchto vrstev přispívá ke vzniku skvrnité makulopatie, ale také ke vzácnějším obrazům ztenčení sítnice a vzniku makulární díry. Pravděpodobně vlivem na výživu a metabolismus sítnice mohou ovlivňovat ostatní vrstvy včetně vrstvy fotoreceptorů (2, 4, 6, 8, 9, 13, 16).
Závěr
Případ vzácné oční manifestace bilaterální makulární atrofie u 13leté dívky upozorňuje na souvislost této retinální afekce s AS a zdůrazňuje důležitost správné diagnostiky přínosné zejména u heterozygotních žen, u kterých je variabilita X vázaného onemocnění velká a přítomnost této retinální či jiné anomálie může podpořit podezření na toto celkové onemocnění a napomoci k celkové diagnostice systémového onemocnění. Je zřejmé, že nález makulární atrofie má vliv na centrální zrakovou ostrost, kterou nelze terapeuticky či jinak ovlivnit (8, 13). Retinální abnormality v souvislosti s AS mohou také představovat vyšší riziko budoucí progrese renálního onemocnění (2, 12).
Do redakce doručeno dne 12. 2. 2014
Do tisku přijato dne 15. 5. 2014
MUDr. Inka Krejčířová, Ph.D.
Dětská oční klinika, PDM, LF MU a FN Brno
Černopolní 9
613 00 Brno
inka.krejcir@seznam.cz
Zdroje
1. Altemir, I. et al.: Reproducibility of optical coherence tomography measurements in children. Am J Ophthalmol, 2013 Jan; 155(1): 171–176.
2. Colville, D., Wang, YY., Tan, R. et al.: The retinal “lozenge” or “dull macular reflex” in Alport syndrome may be associated with a severe retinopathy and early-onset renal silure, Br J Ophthalmol, 2009; 93(3): 383–6.
3. Češka, R., Tesař, V., Dítě, P. et al.: Interna, 1. vydání, Praha: TRITON, 2010, 855, s. 541–542.
4. Dolz-Marco, R., Gallego-Pinazo, R., Francés-Munoz, E., et al.: New macular tomography findings in Alport syndrome. Arch Soc Esp Oftalmol, 2012; 87(2): 55–6.
5. Eriksson, U., Holmstrom, G., Alm, A. et al.: A population-based study of macular thickness in full-term children assessed with Stratus OCT: normative data and repeatability. Acta Ophthalmol, 2009; 87(7): 741–5.
6. Fawzi, AA., Lee, NG., Eliott, D. et al.: Retinal findings in patients with Alport Syndrome: expanding the clinical spektrum. Br J Ophthalmol, 2009;93(12): 1606–11.
7. Hentati, N., Sellami, D., Makni, K., et al.: Ocular findings in Alport syndrome: 32 case studies. J Fr Ophtalmol, 2008; 31 : 597–604.
8. Igami, TZ., Lavezzo, MM., Ferraz, DA., et. al: Unusual macular thickness in Alport syndrome: case report. Arq Bras Oftalmol, 2012; 75(4): 283–5.
9. Mete, UO., Karaaslan, C., Ozbilgin, MK., et al.: Alport’s syndrome with bilateral macular hole, Acta Ophthalmol Scand., 1996;74(1):77–80.
10. Mercé, E., Korobelnik, JF., Delyfer, MN., et al.: A new case of giant macular hole in a patient with Alport syndrome. J Fr Ophthalmol, 2012; 35(8): 573–9.
11. Povýšil, C., Šteiner, I., Dušek, P., et al.: Speciální patologie. 2 vydání, Praha: Galén: Karolinum, 2007, 430.
12. Rheault, MN.: Women and Alport syndrome. Pediatr Nephrol, 2012; 27(1): 41-6.
13. Savige, J., Liu, J., DeBuc, DC., et al.: Retinal basement membrane abnormalities and the retinopathy of Alport syndrome. Invest Ophthalmol Vis Sci., 2010; 51(3): 1621–7.
14. Setälä, K., Ruusuvaara, P.: Alport syndrome with hereditary macular degeneration. Acta Ophthalmol (Copenh),1989; Aug;67(4): 409–14.
15. Spraul, CW., Lang, GE.: Cone dystrophy associated with Alport syndrome. Klin Monbl Augenheilkd, 2000 Sep; 217(3): 194–7.
16. Usui, T., Ichibe, M., Hasegawa, S., et al.: Symmetrical reduced retinal thickness in a patient with Alport syndrome, Retina, 2004; 24(6): 977–9.
17. Xu, JM., Zhang, SS., Zhang, Q., et al.: Ocular manifestation of Alport syndrome. Int J Ophthalmol, 2010; 3(2): 149–51.
18. Zhang, KW., Colville, D., Tan, R., et al.: The use of ocular abnormalities to diagnose X-linked Alport syndrome in children. Pediatr Nephrol., 2008 Aug; 23(8): 1245–50.
19. Zhao, C., Wang, F., Zhang, Y., et al.: A novel splice site mutation in the COL4A5 gene in a Chinese female patients with rare ocular abnormalities. Mol Vis, 2012; 18 : 2205–12.
Štítky
OftalmológiaČlánok vyšiel v časopise
Česká a slovenská oftalmologie

2014 Číslo 3
- Pomocné látky v roztoku latanoprostu bez konzervačních látek vyvolávají zánětlivou odpověď a cytotoxicitu u imortalizovaných lidských HCE-2 epitelových buněk rohovky
- Konzervační látka polyquaternium-1 zvyšuje cytotoxicitu a zánět spojený s NF-kappaB u epitelových buněk lidské rohovky
- Dlouhodobé výsledky lokální léčby cyklosporinem A u těžkého syndromu suchého oka s 10letou dobou sledování
- Syndrom suchého oka
- Cyklosporin A v léčbě suchého oka − systematický přehled a metaanalýza
Najčítanejšie v tomto čísle
- Peeling vnitřní limitující membrány na odchlípené makule jako prevence rozvoje epimakulární membrány po pars plana vitrektomii pro rhegmatogenní amoci sítnice
- Operační řešení těžkého stupně ptózy horního víčka pomocí fronto-tarzálního závěsu trubičkou z biokompatibilního PVC
- Vzácná oční manifestace u podezření na Alportův syndrom
- Konzervační činidla – denní dávka benzalkonium-chloridu v léčbě glaukomu z pohledu pacienta