
Postižení plic u pacientů s multiorgánovou formou histiocytózy z Langerhansových buněk. Popis 8 pacientů a přehled literatury
Pulmonary involvement in patients with multiorgan Langerhans cell histiocytosis. Eight case studies and literature review
Pulmonary Langerhans cell histiocytosis (LCH) manifests with dyspnoea and a cough with no significant expectoration; in some patients, spontaneous pneumothorax is the first symptom. The disease is caused by multiple granulomas in terminal bronchioles, detectable as nodules on high resolution CT (HRCT). During further course of the disease, these nodules progressed through cavitating nodules into thick-walled and, subsequently, thin-walled cysts. LCH may affect the lungs only or multiple organs simultaneously. Pulmonary LCH may continually progress or remit spontaneously. Treatment is indicated in patients in whom pulmonary lesions are associated with multi-system involvement or when a progression of the pulmonary lesions has been confirmed. Our centre has a register of 24 patients with LCH; pulmonary LCH had been diagnosed in 8 patients. The present paper provides information on the course of pulmonary LCH in the individual patients and HRCT images of the pulmonary lesions in these patients.
Key words:
Langerhans cell histiocytosis – pneumothorax – emphysema – 2-chlorodeoxyadenosine – cladribine
Autoři:
Z. Adam 1; T. Nebeský 2; P. Szturz 1; J. Neubauer 3; M. Krejčí 1; L. Pour 1; I. Hanke 4; M. Doubková 5; Z. Merta 5; R. Hájek 1; Z. Řehák 3; R. Koukalová 3; J. Mayer 1
Působiště autorů:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Vlastimil A. Válek, CSc.
2; Oddělení nukleární medicíny a pozitronové emisní tomografie Masarykova onkologického ústavu Brno, přednosta prim. MUDr. Karol Bolčák
3; Chirurgická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Zdeněk Kala, CSc.
4; Klinika tuberkulózy a respiračních nemocí Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednostka prof. MUDr. Jana Skřičková, CSc.
5
Vyšlo v časopise:
Vnitř Lék 2010; 56(Supplementum 2): 105-122
Kategorie:
Histiocytóza z Langerhansových buněk a některá další vzácná hematologická onemocnění
Souhrn
Plicní forma histiocytózy z Langerhansových buněk (Langerhans cell histiocytosis – LCH) se manifestuje dušností a kašlem bez výrazné expektorace, někdy je prvním příznakem spontánní pneumotorax. Nemoc je způsobena vznikem četných granulomů v terminálních bronchiolech, viditelných na CT s vysokým rozlišením (HRCT) jako noduly. V průběhu dalšího vývoje tyto noduly přecházejí přes kavitované noduly do silnostěnných a posléze tenkostěnných cyst. LCH může izolovaně postihovat plíce nebo současně více orgánů. Plicní forma LCH může kontinuálně progredovat anebo spontánně ustoupit. Léčba je indikována v případech, kdy je postižení plic součástí multisystémového postižení nemocného, nebo když je jasná progrese plicního nálezu. Na našem pracovišti registrujeme 24 pacientů s LCH, plicní forma byla diagnostikována u 8 z nich. V článku přinášíme informaci o průběhu plicního postižení u jednotlivých nemocných a HRCT obrazy plicního postižení u těchto nemocných.
Klíčová slova:
histiocytóza z Langerhansových buněk – pneumotorax – emfyzém – 2-chlorodeoxyadenosine – kladribin
Úvod
Plicní forma histiocytózy z Langerhansových buněk (LCH) je vzácné onemocnění. Příznaky plicní formy LCH jsou netypické, kašel, mírné bolesti na hrudníku, případně dušnost a později pneumotorax. Nemoc vzniká ve formě drobných nodularit v plicním parenchymu, které pomalu přecházejí v kavitované nodularity a posléze silnostěnné a slabostěnné cysty. Jejich prasknutí může způsobit pneumotorax. V některých případech může být pneumotorax (vzniklý u mladého člověka) prvním symptomem této nemoci. Uvedené změny struktury plicní tkáně někdy provází i fibrotizace.
Uvedené plicní změny při LCH bývají mylně interpretovány jako plicní emfyzém. Diferenciálně diagnostické znaky emfyzému a plicní formy LCH, viditelné na CT s vysokým rozlišením (high resolution CT – HRCT), rozvádíme v diskuzi.
Diagnostikovat lze tuto nemoc histologickým vyšetřením plicní tkáně odebrané z místa, kde jsou přítomny mikronodularity. Alternativou je bronchoalveolární laváž, avšak vyšetření přítomnosti CD1a pozitivních elementů (Langerhansových buněk) ve vzorku bronchoalveolární tekutiny není dostupné všem pracovištím, která dělají bronchoalveolární laváže.
LCH může být omezená pouze na plicní parenchym, což mívá obvykle lepší prognózu než plicní forma LCH provázená postižením dalších orgánů (mozku, kostí, kůže, lymfatických uzlin).
Po stanovení diagnózy „plicní forma LCH“ stojí lékař před otázkou: Je tato nemoc stále aktivní a mám ji léčit? Nebo mám před sebou již důsledky dříve aktivní LCH a viditelné změny v plicích odpovídají konečné formě dříve aktivní, dnes neaktivní nemoci?
Odpovědi na tyto otázky se hledají velmi obtížně. U pacientů s LCH dochází totiž často v ke spontánním remisím nemoci. Proto samotné stanovení diagnózy není současně indikací k zahájení léčby, terapie je indikována až při průkazu progresivního vývoje nemoci.
Zvyšování četnosti nodularit na kontrolních HRCT plic signalizuje progresi. Jenže kvantifikace nodulů je obtížná. Zvětšování stávajících cyst je doběhem dříve aktivního procesu, a proto tyto změny nelze hodnotit jako progresi, byť jsou dobře na HRCT zřetelné.
Dalším záchytným bodem pro rozhodování o aktivitě, a tedy o indikaci léčby, je funkční plicní vyšetření. Jenže to je velmi hrubý ukazatel a nemusí rozlišit, zda jde o důsledek doběhu „end stage“ změn v parenchymu či současné aktivity nemoci.
HRCT a funkční plicní vyšetření však jsou stále jedinými standardně používanými metodami pro hodnocení aktivity a rozhodování o léčbě. Přínosu PET CT u těchto nemocných se věnuje samostatný článek tohoto supplementa.
Pacienti
Charakteristika souboru pacientů
Na našem pracovišti máme evidováno celkem 24 pacientů s LCH, plicní postižení touto nemocí jsme prokázali u 8 z nich, u 6 byla provedena opakovaná PET-CT vyšetření. Protože do našeho centra jsou odesíláni pacienti dominantně s kostním či multisystémovým postižením, je pochopitelné, že většina našich pacientů s plicní formou LCH měla současně multisystémové postižení. Pouze jeden pacient byl k nám předán ke sledování z plicního pracoviště s izolovanou plicní formou LCH. Informace o těchto pacientech s hodnocením aktivity plicního procesu podáváme ve formě stručných popisů případů. Základní údaje o pacientech jsou uvedeny v tab. 1.

Stručné popisy případů
1. případ, muž narozený roku 1964
Velmi komplikovaný průběh měla LCH u muže narozeného v roce 1964. V roce 1991 (ve 37 letech věku) se u něj postupně začala objevovat nová a nová kostní ložiska. Byl léčen chemoterapií, radioterapií a vysokodávkovanou chemoterapií. Osud tohoto pacienta jsme popsali jinde.
Zde chceme jen zdůraznit, že v průběhu léčby bylo pro neproduktivní dráždivý kašel, námahovou dušnost a nespecifické bolesti na hrudníku provedeno vyšetření plic metodou HRCT, na němž byly patrné četné nodularity a cysty. Výsledek byl kompatibilní s plicní formou histiocytózy, vzhledem k multifokálnímu postižení jsme postižení plic nepovažovali za nutné dále histologicky ověřovat. Plicní postižení bylo zřejmě důvodem k opakovaným plicním infekcím v průběhu chemoterapie i po ní. Aktivitu plicní formy jsme u tohoto pacienta nijak cíleně nevyhodnocovali, protože dominovaly subjektivní potíže způsobené recidivujícími kostními atakami, kterézpůsobily četné patologické frak-tury. Pacienta jsmeztratili ze sledováníroku 2002, kdy byl přijat s další atakou pneumonie na sektorové interní oddělení, kde na tuto infekční komplikaci zemřel. Pitván nebyl.
Tento případ ilustruje, že v případě multiorgánového poškození, které atakuje i plíce, je nutno počítat s vyšším počtem infekčních komplikací v této oblasti.
2. případ, muž narozený roku 1972
U tohoto pacienta vznikla v roce 1993 (ve 21 letech) první ataka LCH, která se projevila bolestí v pravé stehenní kosti a dále v obratli Th12. Osteolytická ložiska byla operována, čímž byla zjištěna diagnóza a léčena operačně alogenními štěpy. Po 10 letech (v roce 2003) nemoc znovu recidivovala. Nově se objevila bolest žeber, páteře. Zobrazovací metody prokázaly nová ložiska na kalvě, velké osteolytické ložisko v 7. žebru vpravo a suspektní ložiska v 4., 5. a 6. žebru. Taktéž bederní obratle byly nově postiženy vícečetnými ložisky. Histologie žebra prokázala recidivu LCH.
Protože zobrazovací metody (RTG snímky, scintigrafie skeletu, CT a MR) prokázaly mnohočetná osteolytická ložiska, přistoupili jsme k systémové chemoterapii. Pacient dostal 5 cyklů chemoterapie (2-chlorodeoxyadenosin 5 mg/m2 s.c. 1.–5. den). Léčba byla ukončena v listopadu roku 2003.
V roce 2007 si nově stěžoval na dušnost a bolest v oblasti hrudního koše. Na provedených zobrazovacích vyšetřeních (scintigrafie skeletu, RTG snímky a PET-CT) nebyla zřetelná progrese ve skeletu. Pro dušnost bylo provedeno HRCT vyšetření, které odhalilo cystická plicní ložiska různé velikosti, místy splývající, ale také četné nodularity i fibrózní proužky. Tyto plicní změny odpovídaly plicní formě LCH (obr. 1–3). Mladý muž podstoupil také bronchoskopii s broncho-alveolární laváží,v níž převažovaly makrofágy. Nebylo však provedeno cytochemické vyšetření přítomnosti buněk exprimujících CD1 antigen a protein S-100 a bez jejich průkazu se nemůže hodnotící vyjádřit k přítomnosti či nepřítomnosti plicní formy LCH, vyšetření však vyloučilo jiné formy plicního poškození. Po edukaci o souvislosti plicní formy LCH a kouření mladý muž přestal kouřit. Subjektivní potíže (kašel a dušnost) se po ukončení kouření zmírnily. Nadále byl kontrolován metodou PET-CT, HRCT, RTG snímky a opakovaným funkčním plicním vyšetřením.



Poslední funkční plicní vyšetření bylo v červnu roku 2009 beze změny.
V červenci roku 2009 bylo provedeno HRTC plic vyšetření se závěrem zcela diskrétní nodularity, zatímco plicní cysty stále přetrvávají beze změn.
Na posledním kontrolním HRCT vyšetření v lednu roku 2010 je zřetelné další zmnožení cyst, jsou lépe ohraničené, tenkostěnné, oproti minulému vyšetření zřetelně odlišitelné od centrilobulárního emfyzému. Nodularity nebyly na HRCT prokazatelné, takže v současnosti plicní formu LCH u tohoto muže považujeme za neaktivní.
3. případ, muž narozený roku 1974
Tento muž (kuřák) byl zdráv až do prosince roku 2007 (33 let věku), kdy u něj byl prokázán diabetes insipidus i deficit dalších hormonů, dominantně androgenů. Na MR mozku byla zřetelná rozšířená stopka hypofýzy s patologickým infiltrátem. Od roku 2007 se také datuje chronický zánět v oblasti obou zvukovodů, který nebyl histologicky vyšetřen, byť je to typický projev LCH. Dalším problémem bylo svědění kůže v oblasti perianální. V rámci diferenciální diagnostiky infiltrace stopky hypofýzy jsme provedli první PET-CT vyšetření (duben roku 2009). V oblasti plicního parenchymu byly prokázány v low dose CT obraze četné drobné cystické útvary různého tvaru, velikosti od 0,8 do 0,9 cm, některé s poměrně dobře vyznačenou stěnou. Současně ojedinělé menší acinární opacity a okrsky zesíleného intersticia. Pleurální prostor bez výpotku. V mediastinu nebyly zvětšené uzliny (obr. 4 a 5). V PET obrazu byla jedině zvýšená kumulace radioaktivní glukózy v oblasti nazofaryngu, ale jen hraniční, jinak nebylo nalezeno nic patologického.
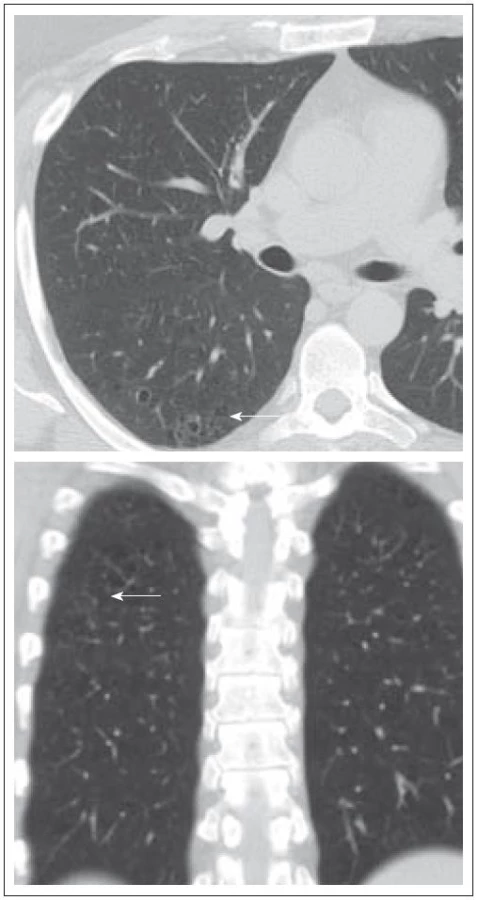

Na HRCT vyšetření provedeném v únoru roku 2009 byly patrné vícečetné shluky drobných cystoidů s vcelku tenkou pravidelnou stěnou, tyto kavitace se nacházejí i samostatně. Výskyt je v relativně typické lokalitě horních laloků a proximálních segmentů laloků dolních. Obraz byl typický pro plicní formu LCH. Na kontrolním vyšetření v odstupu půl roku se nález nemění.
U pacienta byla proto provedena bronchoalveolární laváž s průkazem 10 % CD1pozitivních elementů obsahujících protein S-100. Tento nález odpovídal plicnímu postižení LCH, aktivní nemocí.
Kontrolní HRCT bylo provedeno v říjnu roku 2009, zobrazilo vícečetné drobné cysty do velikosti 8 mm, cysty měly tenkou stěnu. Mimo jedné drobné mikronodularity nebyly žádné další mikronodularity zastiženy. Funkční plicní vyšetření bylo v říjnu roku 2009 bez patologických změn.
V prosinci roku 2009 bylo provedeno kontrolní MR mozku. Při srovnání s předchozími MR z března roku 2008 a prosince roku 2008 bylo patrné, že stále chybí typický hyperintenzivní signál neurohypofýzy. Adenohypofýza měla normální velikost bez zřetelného ložiska. Bobulovitě rozšířené infundibulum se oproti předchozí kontrole zvětšilo o 1 mm z 4,5 na 5,5 mm. Výraznější bylo i sycení ložiska postkontrastně a jeho demarkace vůči proximální části stopky. Jako vedlejší nález popsáno drobné necharakteristické ložisko 3 mm pareventrikulárně vlevo, které dříve nebylo viditelné při MR zobrazení mozku.
Vzhledem k progresi nálezu v oblasti CNS byla v březnu roku 2010 zahájena léčba 2-chlorodeoxyadenosinem.
4. případ, muž narozený roku 1969
Ani tento muž nemá v anamnéze žádné vážnější nemoci. První pneumotorax jej překvapil v roce 2008 (ve 39 letech věku) a od té doby byl sledován na plicní klinice. Pneumotorax s nutností léčebné intervence se celkem 3krát opakoval. Teprve po 3. spontánním pneumotoraxu bylo rozhodnuto o provedení biopsie plic. Ta na základě histologického hodnocení vzorku plicního parenchymu prokázala LCH. V červenci roku 2009 byla provedena kožní excise ze kštice pro makulopapulózní vyrážku. Překvapením byl průkaz kožní formy LCH. Později byla kožní ložiska potvrzena i v intertriginozních oblastech axil, třísel i na hrudníku.
Při HRCT plic v květnu roku 2009 se zobrazily oboustranně v plicních křídlech četné splývané dutiny se zesílenou stěnou 1–2 mm a septy. Maximum těchto změn bylo v horních lalocích pod ventrální stěnou hrudníku a dále difuzně v dolních lalocích. Cystoidy jsou různé velikosti, od několika mm až po 3 cm, mají splývající charakter a vytvářejí obraz hnízd. Bez patrných nodularit v plicních křídlech. Mediastinum nebylo rozšířené a uzliny nebyly zvětšené (obr. 6 a 7).


Na kontrolním vyšetření s odstupem půl roku se rozsah onemocnění zásadně nemění, dochází k splývání cystoidů, dnes spíše tenkostěnných. Nadále jsou šetřeny brániční úhly.
20. května 2009 bylo provedeno PET-CT s nálezem mnohočetných bul s maximem v horních a středních polích. Fluorodeoxyglukóza se akumulovala mírně jedině v oblasti infrahyoidní.
Funkční plicní vyšetření: středně těžká restriktivní ventilační porucha (inspirační vitální kapacita, 2,51 = 47 % náležité hodnoty a TLC – total lung capacity 5,15 = 68 % náležité hodnoty).
V tomto případě jsme měli před sebou pacienta s postižením dvou orgánů, kůže a plic.
V dubnu roku 2009 byla proto zahájena léčba 2-chlorodeoxyadenosinem v kombinaci s cyklofosfamidem a dexametazonem, podáno celkem 6 cyklů léčby, poslední v září roku 2009. Od 4. cyklu byl ke 2-chlorodeoxyadenosinu přidán cyklofosfamid a dexametazon. Došlo ke zmírnění kožních projevů, které byly v oblasti axil, ale i ve vlasaté části hlavy. Reziduální kožní projevy byly vyléčeny metodou PUVA.
Další kontrolní PET-CT vyšetření bylo provedeno v březnu roku 2010. Při low dose CT opět byly zřetelné mnohočetné cystoidní formace v plicním parenchymu, při srovnání s předchozím vyšetřením bylo zřetelné splývání bul ve větší formace.
5. případ, muž narozený roku 1963
Prvním problémem tohoto nemocného byla dušnost, a to taková dušnost, že vedla k hospitalizaci na interním oddělení v roce 2006 (ve 43 letech věku). Nějakou dobu před tím mu začaly vypadávat zuby, příčina nebyla rozpoznána, infiltrace gingivy a někdy i čelisti a uvolňování zubů je také typickým projevem LCH, ale pokud stomatolog o tomto projevu LCH vůbec neví a neodešle kousek tkáně na histologii, nemoc dále pokračuje – tak tomu bylo v tomto případě, příčina vypadávání zubů nebyla zubním lékařem rozpoznána.
Podobně dopadl výsledek vyšetření plicními specialisty, jejich diagnostický závěr – rozedma plic – neodpovídal pravdě.
A tak tento muž chodil na kontroly ke svým spádovým plicním specialistům, aniž by byla LCH u něho lékaři rozpoznána.
Asi od počátku roku 2008 se přidalo mokvání a výtok z ucha. Byl opakovaně ošetřován na ORL ambulanci v místě bydliště a opět bez histologického zkoumání procesu.
Koncem roku 2008 byl předán na ORL kliniku FN u sv. Anny v Brně, kde mu udělali CT vyšetření lební baze. Vlevo bylo zastření zevního zvukovodu s destrukcí jeho dorzální stěny v rozsahu kolem 8 mm s rozpadovou dutinkou v procesus mastoideus velikosti 2 cm, která byla totálně zastřená. Porušeno bylo kostní ohraničení kraniální části svislého úseku canalis nervi facialis. Kostní ohraničení struktur vnitřního ucha včetně polokulovitých kanálků se zdálo být zachovalé. Rozrušena byla i dorzální stěna středouší, ventrální kortika procesus mastoideus a v krátkém úseku i přední hrana pyramidy v sousedství Eustachovy trubice. Stručně řečeno – rozsáhlý destruktivní proces levé pyramidy.
ORL specialisté tak stáli před otázkou: Je to zánět, nebo tumor? Odpověď na tuto otázkou přineslo histologické vyšetření resekátu patologických tkání. Byla to LCH.
V rámci pátrání po rozsahu nemoci byla provedena následující vyšetření:
HRCT plic 17. 3. 2009: difuzní postižení plicního parenchymu – mnohočetné silnostěnné formace bilaterálně o velikosti 3–80 mm, rozložení bylo náhodné, kostofrenické úhly byly šetřeny. Cysty splývaly a byla zřetelná voštinovitá struktura plic s podílem fibrózních změn. Dále byly patrné drobné nodularity v plicním parenchymu a subpleurálně. Bylo zřetelné zesílení nástěnné pleury dorzobazálně bilaterálně (obr. 8).

Při vyšetření scintigrafie skeletu dne 5. 1. 2009 byla aktivita jedině v oblasti levého procesus mastoideus.
V březnu roku 2009 bylo provedeno také PET-CT. Low dose CT vyšetření potvrdilo patologické změny plicního parenchymu a PET zobrazení hraniční akumulaci 18fluorodeoxyglukózy v oblasti levého středního ucha – v defektu po antromastoidektomii.
Kontrolní PET-CT vyšetření bylo provedeno 28. 8. 2009, na něm již nebyla akumulace fluorodeoxyglukózy v oblasti levého ucha a ubylo nodularit v plicním parenchymu.
Spirometrické vyšetření: restriktivní porucha mírného stupně, inspirační vitální kapacita byla 3,66 = 70 % náležité hodnoty, TLC = total lung capacity 4 = 65 % náležité hodnoty.
Léčba: vzhledem k postižení dvou orgánů – plic a ORL oblasti – použita radioterapie na oblasti levého ucha v dávce 20 Gy a celková chemoterapie 2-chlorodeoxyadenosinem v monoterapii 5 mg/m2 5 dní po sobě, celkem 6 cyklů.
Poslední PET-CT kontrola proběhla v březnu roku 2010. Nově v levé dutině čelistní hyperplastické změny šíře 23 mm (sinusitida), stav po antromastoidektomii v oblasti levé pyramidy s pooperačním defektem neměnného charakteru. Low dose CT popsalo stacionární obraz sledovaných kavit v obou plicních křídlech, ale nyní bez ložiskových nodularit v plicích, které byly zřetelné na předchozích snímcích. Při PET zobrazení bez patologické akumulace radiofarmaka. I přes nepřítomnost zřetelné aktivity nemoci však došlo v březnu roku 2010 k opakování pneumotoraxu, který vyžadoval hospitalizaci na chirurgii a torakoskopický zásah.
Na kontrolním HRCT, provedeném v lednu roku 2010, trvaly mnohočetné cystoidní změny plicního parenchymu, v mezidobí se některé cysty lehce zvětšily, jejich charakter byl však stejný. Nadále jsou patrné nepravidelné, ojedinělé, drobné nodularity, jejich množství je oproti minulému vyšetření menší.
6. případ, muž narozený roku 1973
Tento muž byl bez vážnějších chorob až do října roku 2008 (do 35 let věku), kdy si poprvé všiml zvětšených uzlin v oblasti pravého třísla a postupně zjišťoval, že zvětšené uzliny má nejen v třísle, ale také na krku. Měl bolesti v oblasti pravého třísla a bederní krajiny a od listopadu roku 2008 se začaly objevovat undulující teploty dosahují až 38 °C. První uzlina (z nadklíčku) byla exstirpována v listopadu roku 2008 a histologický závěr zněl: histiocytóza z Langerhansových buněk. První návštěva pacienta na našem pracovišti byla v únoru roku 2009. V té době měl generalizované zvětšení uzlin na krku, axilách a v tříslech, velikosti kolem 2 cm.
Dle vstupního PET-CT bylo v CT obrazu zřetelné patologické zvětšení uzlin na krku, v axilách, dále v oblasti dutiny břišní, retroperitoneálně a v malé pánvi, kde byla naměřena největší velikost uzliny 3,5 cm v průměru. Tyto uzliny byly aktivní i při PET zobrazení, aktivita se pohybovala mezi 5,0 SUVmax v axilách a 12,3 SUVmax v oblasti pravých ilických uzlin. Kostní postižení nebylo v té době detekováno dle low dose CT. Při PET-CT vyšetření byly v plicích popsány miliární stíny, tedy známky plicní formy LCH (obr. 9). Klinický obraz zcela jednoznačně svým průběhem odpovídal malignímu lymfomu či lymfogranulomu. S generalizovaným postižením uzlin bez výrazného postižení dalších tkání jsme se zatím u pacientů s LCH nesetkali. Proto jsme diagnózu ověřili odebráním další uzliny z třísla a dále byla provedena biopsie perianální kůže. Jak v tříselné uzlině, tak v perianální kůži byla nalezena infiltrace LCH.

Vzhledem k agresivní formě nemoci jsme se rozhodli pro provedení sběru periferních kmenových buněk po stimulačním režimu obsahujícím etoposid a cyklofosfamid, protože další léčba 2-chlorodeoxyadenosinem by výrazně snížila vyhlídky na úspěšný sběr. Ihned po podání této chemoterapie ustaly teploty a došlo k výraznému zmenšení uzlin. Pak následovala chemoterapie 2-chlorodeoxyadenosin 5 mg/m2 5 dní po sobě, cyklofosfamid 300 mg 5 dní po sobě a solumedrol 250 mg, celkem 6 cyklů, chemoterapie byla ukončena dne 26. 8. 2009. Pro vyhodnocení účinnosti léčby bylo po 4 cyklech provedeno v pořadí 2. PET-CT vyšetření. Bylo konstatováno, že ubylo miliárních stínků na plicích a dále lymfatické uzliny již byly normální velikosti. V PET obraze došlo k normalizaci původně zvýšené akumulace fluorodeoxyglukózy. Další kontrolní PET-CT vyšetření mělo být s odstupem za delší dobu až po ukončení léčby.
Jenže 5. 11. 2009 byl pacient přijat pro akutní zhoršení stavu (dušnost, teploty) k diferenciální diagnostice, zda se jedná o infekční či neoplastický původ těchto potíží. V pořadí 3. PET-CT vyšetření prokázalo v low dose CT obrazu stále nezvětšené lymfatické uzliny, novinkou však byla osteolytická ložiska v kostech kyčelních, dříve nedetekovaná. PET vyšetření však již odpovídalo relapsu nemoci v doposud nezvětšených uzlinách a nově byl zřetelný na PET i CT obraze relaps v kyčelních kostech (obr. 10).

Současně bylo provedeno také HRCT vyšetření, kde byly popsány četné drobné nodularity, místy se již u některý uzlů vytvořily kavitace, a to v horních a středních lalocích. Kostofrenické úhly byly šetřeny. Patologická infiltrace pánve byla potvrzena i na MR obrazu.
Pro velmi časnou recidivu nemoci jsme tedy zahájili chemoterapii CHOEP.
Pro časné vyhodnocení léčebné účinnosti této chemoterapie bylo provedeno po 2 cyklech kontrolní PET-CT vyšetření (26. 1. 2010). Low dose CT vyšetření zobrazilo přetrvávající mnohočetné nodularity v oblasti plic do 8 mm, které nabývají cystoidní vzhled. Ve srovnání s vyšetřením z listopadu roku 2009 byla mírná progrese velikosti opacit. V kostech kyčelních trvají osteolytické defekty. V PET zobrazení již nebyla zřetelná žádná ložiska patologického hypermetabolizmu, která byla zřetelná na předchozím PET zobrazení. To signalizuje chemosenzitivitu nemoci. V březnu 2010 pacient ukončil léčbu vysokodávkovanou chemoterapií BEAM s autologní transplantací a kontrolní PET-CT vyšetření bylo bez patologické akumulace fluorodeoxyglukózy.
U tohoto pacienta jsme v první fázi věnovali pozornost uzlinové formě nemoci, plicní postižení nedominovalo. Nicméně již při 1. vyšetření v lednu roku 2009 byly na HRCT četné nodularity, takže aktivní plicní choroba (obr. 11).

7. případ, muž narozený roku 1963
I tento muž byl relativně zdráv až do března roku 2008 (do 45 let věku). Pro bronchitidu byl přeléčen antibiotiky a pro přetrvávající kašel odeslán na plicní oddělení, kde po RTG snímku plic provedli následně HRCT a diagnostikovali mikronodulární rozsev do 5 mm bilaterálně. Následně byla provedena u tohoto nemocného bronchoalveolární laváž. Z cytologického vyšetření získané tekutiny bylo konstatováno, že jsou přítomny s velkou převahou makrofágy a téměř všechny elementy jsou protein S-100 pozitivní, CD1 pozitivní, což svědčí pro Langerhansovy buňky.
Tímto vyšetřením byla potvrzena diagnóza. Pacient, kuřák, přestal po vysvětlení kouřit.
V červenci roku 2008 bylo provedeno PET-CT, na němž nebylo nic patologického popsáno. Podobný výsledek byl při kontrolním vyšetření v únoru roku 2010 – bez jakéhokoliv nového suspektního ložiska, při low dose CT zobrazení byly na plicích patrné pouze drobné cysty, nikoliv však nodularity. A dle funkčního vyšetření plic nedošlo k žádné podstatné změně.
8. případ, žena narozená roku 1953
Tato pacientka je předmětem samostatného case reportu, v němž jsou i obrazy plicního postižení. Vzhledem k tomu, že klasický snímek plic byl dlouhodobě nenápadný a tato paní si na plicní problémy nestěžovala, byla plicní forma LCH zjištěna až při závažné pneumonii, která způsobila dušnost a hypoxemii. Podrobnosti o průběhu, HRCT a RTG obrazy jsou uvedeny v samostatném case reportu tohoto supplementa popisujícím ORL manifestaci LCH.
Diskuze
Maligní nemoci odvozené od histiocytů jsou podstatně vzácnější než nemoci odvozené od lymfoidních či od myeloidních buněk. WHO klasifikace krevních chorob z roku 2009 popisuje mimo LCH také sarkom z Langerhansových buněk [32], který může případně vznikat z LCH ložisek [33].
Etiologie LCH není jasná a odbornou literaturou se táhla dlouho diskuze, zda jde o aberantní imunologickou reakci či zda jde o maligní klonální onemocnění. Klonální původ byl prokázán již v roce 1994 [35–37]. Nicméně jasné také je, že zánětlivé procesy indukují pomnožení Langerhansových buněk [38,39] a epidemiologicky je jasná souvislost mezi kouřením a plicní formou LCH. V současnosti někteří autoři považují LCH za aberantní reakci imunitního systému, zatímco jiní za klonální maligní onemocnění.
Biologie Langerhansových buněk
Langerhansovy buňky patří do linie dendritických buněk. Zralé dendritické buňky mají heterogenní původ. Mají sice společný původ v hemopoetických kmenových buňkách, z nich se ale mohou vyvíjet cestou lymfoidního nebo myeloidního prekurzoru. Společnou funkcí všech dendritických buněk je prezentovat antigen T buňkám.
Myeloidní dendritické buňky (DC1) mají původ v CD34 pozitivním myelomonocytárním prekurzoru, z něhož vznikají za spoluúčasti granulocyte macrophage stimulating factor – GM-CSF a tumor nekrotizujícího faktoru α (TNF α). Tyto buňky lze také získat z monocytů izolovaných z krve působením GM-CSF a interleukinu-4. Z těchto prekurzorů mohou vzniknout také Langerhansovy buňky, zvláště v přítomnosti transforming growth factor β, který má zásadní význam pro diferenciaci směrem do Langerhansových buněk [40–44].
Později byly identifikovány další populace dendritických buněk, které se odvozují z buněk lymfatické linie pod vlivem interleukinu-3. Za určitých podmínek i tyto buňky mají potenciál se transformovat a dát vznik dendritickým buňkám [45].
Na rozdíl od dendritických buněk, které jsou přítomny téměř ve všech tkáních, jsou Langerhansovy buňky lokalizovány specificky v epidermis, v mukózních površích, včetně mukózy dýchacích cest. Langerhansovy buňky se liší od ostatních dendritických buněk přítomností specifických organel v jejich cytoplazmě – Birbeckových granul, která jsou zřetelná pouze v elektronovém mikroskopu. Birbeckova granula se podílejí na internalizaci exogenních substancí.
Langerhansovy buňky lze detekovat také na základě exprese specifických markerů (langherin – CD207), což je lektin, který je asociován s Birbeckovými granulemi.
V normální plicní tkáni tvoří Langerhansovy buňky síť v tracheobronchiální výstelce. V oblasti alveolů se normálně vyskytují velmi zřídka. V alveolech se jejich přítomnost zvyšuje u kuřáků a dále v době zánětu. Také v plicních tumorech je jejich počet zvýšen. Jejich počet souvisí s lokální tvorbou GM-CSF [46,47].
Langerhansovy buňky, podobně jako jiné dendritické buňky, mají úlohu strážce. Tyto buňky internalizují antigeny a pak ve spolupráci s proinflamatorními signály putují do lymfatických uzlin, kde stimulují specifickou T buněčnou odpověď [48].
Morfologické projevy plicní LCH
Typickým projevem je akumulace aktivovaných Langerhansových buněk do granulomů, které se tvoří ve stěně distálních bronchiolů a později ji destruují. V těchto granulomech jsou však také přítomny lymfocyty a zánětlivé buňky včetně eozinofilů a makrofágů. Morfologicky se plicní ložiska podobají ložiskům této nemoci v jiných tkáních. Na rozdíl od ostatních tkání jsou plicní ložiska vždy mnohočetná, ale drobná, tvoří nodularity obvykle 3–6 mm v průměru.
Při vyšetření takové nodularity jsou ve světelném mikroskopu zřetelné Langerhansovy buňky nepravidelných tvarů, průměrné velikosti 15 µm. Mají stočené nepravidelné jádro a světle eozinofilní cytoplazmu, která neobsahuje žádné anebo ojedinělé fagocytované částice. Imunohistochemickým barvením je možné prokázat, že se jedná opravdu o Langerhansovy buňky. Používá se k tomu protilátka proti antigenu CD1a a někdy také průkaz langerinu, což je lektin specificky exprimovaný těmito buňkami. Specifické barvení proteinu S-100, dříve velmi často používané, není příliš specifické, tento protein je prokazován také na neuroendokrinních buňkách a na některých makrofázích.
V elektronovém mikroskopu je možná vizualizace Birbeckových granul, která jsou v patologických Langerhansových buňkách četnější než ve fyziologických Langerhansových buňkách [49–52].
Výše uvedená ložiska jsou v plicní tkáni difuzně rozprostřena, jsou neostře ohraničená od okolní zdravé tkáně. LCH granulomy dominantně postihují stěnu centrálních a terminálních bronchiolů, kterou postupně destruují. Z tohoto hlediska plicní forma LCH připomíná spíše bronchiolitidu než difuzní plicní postižení. Granulomy jsou neohraničené a dosahují do sousedních alveolárních struktur. Tyto alveoly pak obsahují zvýšené množství makrofágů s pigmentem a připomínají deskvamativní intersticiální pneumonii [53,54].
V těch částech plic, které nejsou postiženy, je normální histologický obraz plicní tkáně, obvykle s výjimkou změn způsobených kouřením. Někdy však je LCH spojena se vznikem difuzní plicní fibrózy. Nevyjasněnou otázkou je, proč tato fibróza u některých nemocných je a u jiných není přítomna.
Ložiska plicní formy LCH podléhají stejným změnám jako jiná ložiska v těle. Zpočátku je v nich hodně Langerhansových buněk, v pozdějších fázích přibývá eozinofilů a dalších zánětlivých buněk. V terminálních fázích vývoje tohoto ložiska jsou přítomny pouze ojedinělé Langerhansovy buňky nebo žádné. Makroskopický vzhled se také mění, z původního nodulu vzniká kavitovaný nodulus a posléze cystický úvar s nepatrným vazivovým lemem [48].
Epidemiologie plicní formy LCH
Plicní forma LCH u dospělých je vzácným nálezem. V některých případech může jít o projev nemoci omezený na plicní parenchym. V těchto případech se používá termín izolovaná plicní forma LCH. V jiných případech je postižení plic součástí vícesystémové formy LCH.
Izolovaná plicní forma LCH se vyskytuje dominantně u kuřáků. Incidence LCH s postižením plic není přesně zmapována. Dostupné jsou pouze relativní údaje, popisují podíl pacientů s plicní formou LCH mezi všemi nemocnými s intersticiálním plicním postižením (tab. 2). Rozsáhlejší epidemiologická data jsme našli z Japonska, kde udávají prevalenci u mužů 0,27/100 000, u žen 0,07/100 000 [55].

Stejně tak není přesně známa incidence všech forem LCH. Starší data uvádějí 1–2 případy LCH v dospělosti na 1 000 000 obyvatel. Jasné je, že výskyt LCH v populaci bude vyšší než udávaná incidence 1–2/1 000 000, protože toto onemocnění není často správně diagnostikované, pokud již způsobuje závažné symptomy. Přitom mnozí pacienti s LCH se ani k lékaři nedostaví, protože nepociťují žádné symptomy, anebo symptomy jsou jen dočasné, nemoc u nich dospěje ke spontánní remisi.
Častější používání vysoce rozlišovacího CT zobrazení (HRCT) může přispět k častějšímu rozpoznávání této plicní manifestace LCH.
Plicní forma LCH nejčastěji postihuje mladé osoby ve věku 20–40 let a postihuje v 90 % kuřáky.
Plicní forma LCH je diagnostikována po radioterapii či chemoterapii maligních lymfomů či Hodgkinova lymfomu [56–58], ale také v průběhu onemocnění solidními tumory či při monoklonální gamapatii nejistého významu (MGUS). Další maligní choroby či MGUS se u pacientů s LCH vyskytují častěji než v průměrné populaci. To svědčí o alteraci imunity u pacientů s LCH a také o vhodnosti hledat při ambulantních kontrolách těchto pacientů nejen odpověď na otázku, zda nedochází k recidivě LCH, ale také odpověď na otázku, zda se neobjevují klinické a laboratorní známky dalšího maligního onemocnění.
Příznaky
Ačkoliv se jedná o difuzní plicní postižení, nemocní si často nejsou vědomi žádných potíží (nemoc se rozvíjí pomalu) nebo příznaky dávají do souvislosti s kouřením. Udává se, že při 20–30% postižení plicního parenchymu nodularitami a cystami LCH zůstávají výsledky funkčního plicního vyšetření v mezích normy, zřejmě díky značným kompenzačním mechanizmům plic. To znamená, že při postižení plic tohoto rozsahu nemusí být přítomny žádné plicní symptomy. To potvrzuje 3. popsaný případ, tento muž měl jasné HRCT známky plicní formy LCH, aktivita byla ověřena z bronchoalveolární laváže a přitom neměl vůbec žádné příznaky postižení plic a výsledky plicního funkčního vyšetření měl v normě.
Zahraniční prameny udávají, že asi 25 % případů je odhaleno náhodným snímkem plic, provedeným z jiného důvodu, než je podezření na plicní formu LCH.
K lékaři nemocné přivádí některý z následujícího výčtu možných příznaků:
- suchý dráždivý kašel,
- dušnost při námaze,
- dušnost či kašel ve spojení s typickými B symptomy (subfebrilie či febrilie, noční pocení, úbytek hmotnosti),
- spontánní pneumotorax a bolest jím vyvolaná je příznakem vedoucím ke stanovení diagnózy asi u 10–20 % osob s plicní formou LCH.
Spontánní pneumotoraxy se nejčastěji objevují u mladých mužů [62–65]. Pneumotoraxy se mohou opakovat, jak demonstrujeme dvěma případy v našem souboru.
Hemoptýza není typickým příznakem nemoci, v případě hemoptýzy je nutno pečlivě vyloučit jiné diagnózy, než se uzavře původ hemoptýzy v plicní formě LCH [66].
Některé zahraniční prameny uvádějí, že plicní forma LCH u dospělých je obvykle onemocnění omezené na plicní parenchym a že průkaz postižení jiných tkání je spíše výjimečný (osteolytická ložiska do 20 %, diabetes insipidus do 5 % a případně kožní projevy nemoci [67–69].
V našem souboru však máme pouze 1 pacienta, u něhož byla prokázána primárně plicní forma plicní LCH, ostatní naši nemocní mají multisystémovou formu onemocnění. V tomto případě bude zásadním způsobem záležet na úhlu pohledu autora textu. Pokud text vychází z pera pneumologů, je pochopitelné, že ti se budou setkávat hlavně s plicní formou LCH. Naopak, pokud text vychází z pera hematologů, tak k nim přicházejí pacienti dominantně s jinou než plicní formou LCH a postižení plic se zjistí až po vyšetřeních, která má za cíl stanovit rozsah nemoci.
Projevy plicní formy LCH na klasickém RTG snímku plic
Na standardním RTG snímku plic jsou projevy této nemoci málo zřetelné. Nejčastějším projevem je retikulomikronodulární infiltrace. Někdy mohou být na snímku plic zřetelné cysty. Změny postihují typicky horní a střední plicní pole, ubývá jich směrem bazálním a často vynechávají kostofrenické úhly [70]. Tato distribuce LCH se vysvětluje tím, že horní a střední plicní laloky jsou více ventilované než bazální části plic, a tedy že více exogenních faktorů je vzduchem dopraveno do horních a středních částí plic než do bazálních plicních oblastí. Naopak, bazální části plic jsou více prokrvené než horní, a tak hematogenní metastázy jsou častěji detekovány v dolních plicních polích.
Infiltráty jsou velmi často mnohočetné, i když pacient nemá velké příznaky, ale jsou popsány i výjimky s izolovaným nodulem [71].
Plicní forma LCH není spojena s výpotkem či mediastinální lymfadenopatií až na naprosté výjimky [72].
V případech, kdy změny plicního parenchymu způsobené samotnou LCH nebo LCH a jí indukovanou plicní fibrotizací způsobí plicní hypertenzi, bývají zřetelné výrazné plicní hily.
Při pokročilé formě nemoci mohou postupně mizet plicní nodularity a dominující abnormalitou na RTG snímku jsou cysty, jež mnohdy vypadají jako plíce při emfyzému. Asi v 10 % případů však na běžném RTG snímku nemusí být nic patologického zřetelné.
Projevy plicní formy LCH v CT obraze s vysokým rozlišením (high resolution CT – HRCT)
HRCT zobrazení přineslo zásadní zlom v diagnostice plicní formy LCH. Nyní je základním vyšetřením, které by se mělo provést při podezření na tuto či jinou intersticiální plicní nemoc. HRCT přináší podrobné informace o základních plicních změnách, typických pro tuto nemoc, jimiž jsou noduly, kavitované noduly a cysty, které nemusí být zřetelné na klasickém RTG snímku [73–79]. Při HRCT zobrazení je možné vidět, že retikulace, zřetelná na běžném RTG snímku, je při HRCT zobrazení tvořena malými plicními cystami.
Typickými projevy plicní formy LCH jsou malé, špatně ohraničené noduly, které se postupně vyvíjejí v kavitované noduly a následně v silnostěnné a později tenkostěnné plicní cysty.
Tyto změny postihují jak periferní, tak centrální části plicního pole. Ložiska jsou fokální, jsou oddělena parenchymem, který na HRCT má normální obraz. Nodularity jsou ponejvíce v horním a středním plicním poli a mají tendenci šetřit dolní části plic. Distribuce nodulů je centrilobulární, odráží tak vazbu této nemoci na bronchioly.
S pokračující nemocí pak i na HRCT dominují cystická ložiska. Jejich velikost obvykle nepřesahuje 1 cm, ale cysty mohou postupně splývat, a vytvářet tak vetší konglomeráty – dutiny.
Kontinuální HRCT sledování u vybraných nemocných prokázalo, že radiologické změny se vyvíjejí od nodulu přes kvitovaný nodulus, silnostěnné po slabostěnné cysty. Tato série HRCT snímků také prokázala, že noduly nebo kavitované noduly mohou vymizet, zatímco cysty již jsou definitivní (end stage disease) změnou [80–82].
V diferenciální rozvaze jsou plicní cystické změny v některých případech velmi těžko odlišitelné od centrilobulárního emfyzému. Distribuce je stejná, tedy centrilobulární a chybí zřetelně vyjádřená stěna. Sledování změn v časové ose může pomoci při odlišení obou entit. Někdy však mohou diferenciální rozpaky v HRCT obraze přetrvávat.
Dalšími popsanými změnami u LCH jsou lineární denzity a pak také emfyzematózní buly kuřáků. U některých nemocných je zřetelné rozšíření stínu plícnice [83]. HRCT je přínosem pro rozpoznání intersticiálních plicních procesů nejen u dospělých, ale také u dětí [84,85].
HRCT má zásadní přínos pro určení místa vhodného pro plicní biopsii.
Plicní funkce při LCH
Změny plicních funkcí odrážejí míru anatomických změn a trvání nemoci. Za nejčastější abnormalitu je uváděna redukce difuzní kapacity plic pro CO. Bývá přítomna u 70–90 % nemocných. U velké části nemocných se udává také obstrukční porucha (jak jinak, jde současně o osoby závislé na kouření se značnou expozicí). Dále bývá nízká vitální kapacita (VC) a i mírně snížená celková plicní kapacita plicní (TLC) a zvýšený reziduální objem. Poměr RC/TLC bývá často zvýšený. Hodnota usilovného výdechu bývá snížena u 20–30 % vyšetřovaných a v průběhu nemoci se dále zvyšuje. Tato obstrukční porucha zrcadlí predominantně bronchiolární lokalizaci plicní formy LCH a zároveň také často chronickou kuřáckou bronchitidu. Proto Tazi uvádí, že obstrukční porucha spolu s difuzními plicními infiltráty je velmi pravděpodobně způsobena plicní formou LCH. Nemusí to však být vždy, někteří pacienti trpí čistou restriktivní poruchou [86–93].
Jak jsme již ale dříve uvedli, tak počáteční změny v plicním parenchymu, jasně zřetelné na HRCT, ještě nevedou k patologických výsledkům funkčních plicní testů ani k subjektivním plicním potížím.
Bronchoskopické nálezy a přínos bronchoalveolární laváže pro stanovení diagnózy LCH
Bronchiální strom je při klasickém bronchoskopickém vyšetření normální, nebo jeví pouze nespecifické známky zánětu způsobeného kouřením. Bio-psie bronchiální mukózy není nijak přínosná pro stanovení této diagnózy.
Transbronchiální biopsie může být ve výjimečných případech přínosem, pokud je získán dostatečně velký počet vzorků a všechny jsou kompletně imunohistochemicky přešetřeny [93–95]. Ve větší míře byla transbronchiální punkce používána v USA, diagnostickým přínosem však byly pouze v 10–40 % případů. Tento nízký výtěžek je způsoben fokální distribucí ložisek v parenchymu [93–95].
Přínosnou metodou je však bronchoalveolární laváž, základní poznatky o této metodě uvádí Mayer [96].
Počet buněk v aspirátu z bronchoalveolární laváže je obvykle zvýšený, obvyklé jsou počty buněk nad 106/ml se zřetelnou predominancí alveolárních makrofágů. Zvýšená buněčnatost aspirátu z bronchoalveolární laváže je způsobena kouřením cigaret [97]. Počty alveolárních makrofágů bývají normální u nekuřáků.
Diferenciální rozpočet ve vzorku bronchoalveolární laváže může vykazovat mírný nespecifický vzestup počtu eozinofilů, obvykle méně než 10 % [67,97]. Počet lymfocytů bývá normální či mírně snížený a poměr CD4/CD8 buněk je snížený, což je obvyklé u kuřáků.
Identifikace Langerhansových buněk mezi všemi buňkami získanými bronchoalveolární laváží je možná s pomocí monoklonální protilátky proti CD1a.
Zvýšené počty CD1a pozitivních buněk do 3 % jsou časté u kuřáků, aniž by u nich byla zřetelná plicní forma LCH. Kouření zvyšuje počet Langerhansových buněk v plicích [98]. Mírné zvýšení CD1a pozitivních buněk bylo také zjištěno u pacientů s difuzní intersticiální plicní chorobou, spojenou s alveolární hyperplazií (zvláště u difuzní intersticiální plicní fibrózy) a také v adenokarcinomech plic [67,99–102].
Pokud se použije jako hranice pozitivity nález > 5 % CD1a+ buněk, je již specifika tohoto testu velmi vysoká, ale senzitivita zůstává nízká, 25 % [67,103–105].
Tazi [67] uvádí, že u většiny dospělých s plicní formou LCH je počet CD1a pozitivních buněk nižší než uváděná hranice 5 %, podobně jako je tomu u kuřáků. Pokud bronchoalveolární laváž prokáže dostatečně zvýšený počet CD1a pozitivních buněk a HRCT má odpovídající obraz, tak je možné diagnózu uzavřít jako plicní formu LCH. Pokud však HRCT odpovídá plicní formě a v laváži není dostatečný počet CD1a pozitivních elementů, je nutno přikročit k dalším diagnostickým krokům – plicní biopsii z oblasti s nejvyšší četností nodularit. Podobně ale i v případě, kdy HRCT neodpovídá plicní formě a laváž vyjde pozitivní. V roce 2010 lze mezi odborníky na plicní formu stále najít jak názory, že diagnostika pomocí laváže je zcela dostačující, tak i názory, že pro diagnostiku laváž nestačí a je nutno udělat plicní biopsii.
Domníváme se, že v případech jasného HRCT nálezu by měl být pozitivní výsledek laváže ke stanovení diagnózy plicní formy LCH dostačující.
Stanovení diagnózy
Stanovení diagnózy je založeno na rozpoznání typického LCH granulomu s průkazem dendritických Langerhansových buněk, obsahujících Birbeckova granula a odpovídající CD antigeny (CD1a) [106,107]. Histologický vzorek se klasicky získává torakoskopicky z míst, kde je dle HRCT nejvíce granulomových ložisek. Vzhledem k tomu, že granulomy tvoří disperzní proces, je nutné vzít odpovídající kus tkáně a také jej celý vyšetřit metodou imunohistochemického barvení [108,109].
Pokud je však HRCT plic typické pro tuto nemoc, je na zvážení zkušeného plicního odborníka, zda provádět či neprovádět plicní biopsii. Pokud je na HRCT typická nodulárně cystická struktura plicní tkáně, jedná se o mladého kuřáka a přitom bronchoalveolární laváž prokazuje vysoký počet CD1a+ elementů, není nutné obvykle ověřovat plicní formu LCH pomocí histologie.
Nicméně diferenciálně diagnosticky je nutné vědět o dalších nemocech, které mohou mít podobný obraz (mykobakteriální infekce, Wegenerova granulomatóza, kavitované plicní metastázy, alveolární plicní karcinom, septické emboly).
U dospělých může být obtížné odlišit diferencovanou formu lymfangioleiomyomatózy. V tomto případě však by sonografie a CT měla prokázat angiomyolipomy i v jiných částech těla [110].
Pokud je plicní forma LCH součástí multisystémového postižení, je možné ověřit diagnózu histologickým vyšetřením vzorku odebraného z dostupnějšího patologického ložiska v jiné tkáni či orgánu, než jsou plíce (z kosti, případně z periodontálních ložisek, imitujích zánět dásní apod.) [111,112]. V dospělosti může být kombinována např. plicní forma s postižením stopky hypofýzy a diabetes insipidus [113]. Zde naopak průkaz, že v plicích jsou noduly LCH, osvětlí, co je příčinou infiltrátu v oblasti stopky hypofýzy.
Klinický průběh a prognóza
Klinický průběh je velmi divergentní a nelze jej u konkrétního pacienta předpovědět [67,78]. Asi u 50 % nemocných má izolovaná plicní forma LCH příznivý průběh, k vymizení aktivity nemoci – vymizení plicních opacit a nodularit – dojde buď spontánně, nebo při kortikoidní léčbě. Nicméně pokud dojde k poškození plic formou cyst, tak to je ireverzibilní. Výsledky vyšetření plicních funkcí mají pak podobu setrvalé či progresivně se zhoršující respirační poruchy.
Asi u 20 % nemocných se objevují ihned z počátku nemoci závažnější problémy typu opakujících se pneumotoraxů anebo progresivní respirační selhání se vznikem chronického cor pulmonale.
Asi u 30–40 % nemocných jsou příznaky nemoci dlouhodobé, s postupnou konverzí nodularit na silnostěnné a posléze tenkostěnné plicní cysty. Je velmi obtížné rozlišit, zda jsou přítomny pouze klidné cysty nebo zda se v plicním parenchymu stále tvoří nové nodularity jako projev aktivní nemoci. Sledování nemoci je velmi obtížné, je nutné skládat dohromady výsledky všech dostupných metod, které mohou poskytnout informaci o aktivitě plicní nemoci (funkční plicní vyšetření, HRCT, PET-CT vyšetření) [85,114–122].
Literatura uvádí následující nepříznivé prognostické faktory:
- nástup nemoci ve starším věku,
- delší doba aktivních příznaků nemoci,
- opakované pneumotoraxy,
- mimohrudní projevy nemoci (vyjma kostního postižení, které obvykle nesignalizuje horší prognózu), difuzně rozprostřené plicní cysty,
- závažné poruchy ventilace [90,93,123].
Závažná plicní hypertenze signalizuje špatnou prognózu [124,125].
Plicní forma LCH však znamená také vyšší riziko plicního karcinomu, zvláště u kuřáků, ale také zvýšené riziko ostatních malignit [57,120,126–129].
Léčba
Zásadní důležitost pro vymizení této nemoci má ukončení kouření. Pokud ustane kouření, tak v mnoha případech byla popsána remise nemoci [117,118,130].
O léčbě se uvažuje v případech, kdy po přerušení kouření je zřetelná progrese nemoci. U mnoha pacientů s LCH dochází ke spontánní úpravě nebo jsou stabilní po delší dobu, takže se k léčbě vůbec nepřistoupí [131].
Efektivitu léčby je obtížné vyhodnotit. Odpovědět na otázku, zda se pořád tvoří další a další nodularity, nepřináší snímek plic a nelze očekávat ani od HRCT přesnou odpověď na tuto otázku, i když HRCT je nejdůležitějším prostředkem k hodnocení léčebné odpovědi [119]. Funkční plicní vyšetření odráží hlavně end stage změny. Zhoršování funkčního vyšetření jistě odráží progresi nemoci, regresi nemoci však nelze funkčním vyšetřením potvrdit. Dále počty případů jsou nízké, takže léčebné postupy zde nevyhodnocují žádné prospektivní randomizované studie. Poučení o možnostech léčby je nutné proto extrahovat z popisů malých skupin pacientů či z popisů jednotlivých případů.
Zcela výjimečná je asi léčba pomocí transplantace plic [132,133], protože může dojít k recidivě nemoci v postižené plíci [134–136].
Glukokortikoidy
Pro izolovanou plicní formu LCH ve stadiu nodulárních infiltrátů je nejčastěji používána glukokortikoidní léčba. Obvyklá dávka je 0,5–1,0 mg/kg/den, která se pomalu snižuje v průběhu 6–12 měsíců [67,77,137]. Od glukokortikoidní léčby se očekává, že zabrzdí aktivitu nemoci a potlačí zánětlivou reakci v LCH granulomech, a tím zabrzdí destrukci bronchiolů vedoucí ke tvorbě cyst.
Cytostatické léky
Cytostatické léky (vinblastin a méně často metotrexát nebo 2-chlorodeoxy-adenosin) jsou považovány za indikované v případech, kdy plicní formu LCH provází multisystémové postižení.
Pro případy izolované plicní formy LCH není totiž průkazu, že by tyto léky byly účinnější než pouhé glukokortikoidy. Z druhé strany v případech progrese při glukokortikoidech mají jistě své oprávnění i v léčbě izolované plicní formy LCH.
Prvním cytostatikem, u něhož byl prokázán požadovaný léčebný účinek, byl derivát vinka alkaloidu – vinblastin. Později se do léčby dostal metotrexát, merkaptopurin. O něco později se do léčby dostal další účinný lék, etoposid, který je účinný u plicních i mimoplicních forem [138–140]. Z cytostatik objevených až ke konci minulého století to byl právě 2-chlorodeoxyadenosin, který prokázal svou účinnost u multisystémového plicního postižení. Pozitivní dopad léčby 2-chlorodeoxyadenosinem na plicní formu LCH byl doložen v několika publikacích [141–145].
V případě kožního a dalšího multisystémového postižení lze použít kombinovanou léčbu – PUVA, prednison, merkaptopurin a vinblastin [146].
Další léky
Efekt byl popsán také po etanerceptu [147] a opakovaně po thalidomidu [148].
Z druhé strany nutno zmínit, že byla popsána reaktivace nemoci po léčbě kolitidy a infliximabem [149].
Do komplexní léčby však patří také zvládání infekcí, které se v poškozených plicích často vyskytnou, a dále operační řešení pneumotoraxů či řešení pneumotoraxů pleurodézou [116].
Stanovení rozsahu nemoci a vyhodnocování účinnosti léčby
Největším problémem je vyhodnocování aktivity nemoci, a tedy i efektivity léčby. Základním vyšetřením byly snímky skeletu detekující kostní ložiska a také scintigrafie skeletu. Měkkotkáňovou expanzi lze zachytit pomocí magnetické rezonance (MR) a vyšetřením pomocí počítačové tomografie (CT).
V literatuře se objevilo několik zmínek o přínosu scintigrafického zobrazení aktivních ložisek LCH pomocí oktreotidového skenu [150–152]. Tyto metody však nedošly širšího použití. Přehled všech metod používaných k detekci LCH uvádějí četní autoři [32,153–155,157,159,160,169,170]. Významu PET-CT pro plicní zobrazení je věnován samostatný článek tohoto supplementa.
Závěr pro praxi
- Diagnostika a léčba LCH vyžaduje obvykle spolupráci lékařů více oborů [71].
- U mladých pacientů se závislostí na cigaretách (kuřáků) s dlouhodobějším kašlem a případně dušností je nutno myslet na intersticiální plicní postižení a v rámci diferenciální diagnózy také na plicní formu LCH.
- Včasný průkaz plicní formy LCH a včasná léčba mohou vést k zastavení procesu, který jinak progresivně destruuje plíce.
- Změny v plicním parenchymu způsobené plicní formou LCH jsou velmi dobře viditelné na HRCT plic, zatímco na klasickém RTG snímku nemusí být zřetelné žádné patologické změny. Proto pro vyhodnocování dynamiky plicní formy LCH se standardně používá HRCT vyšetření a funkční plicní vyšetření.
Tato publikace byla připravena v rámci projektu MUNI/A/1012/2009 s názvem „Optimalizace diagnostiky a terapie maligních chorob a komplikací, které tyto maligní nemoci provázejí, s využitím nových molekulárně biologických metod“, a také je součástí aktivit v rámci grantů IGA MZ: NR9225, NS10387 a NS10406.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 9. 9. 2010
Zdroje
1. Horáček J. Primary pulmonary histiocytosis X. Cesk Patol 1975; 11 : 210–213.
2. Bittenglova R, Pešek M, Mukenšnabl P et al. Granulomatóza z Langerhansových buněk. Stud Pneumol Phthiseol 2002; 62 : 196–202.
3. Polák J, Fuchs B. Pulmonary histiocytosis X. Cesk Radiol 1987; 41 : 289–300.
4. Rožánek P, Molnar V, Rešl M. Tři případy plicní granulomatózy z Langerhansových buněk. Lék Zpr Lék Fak Univ Karlovy Hr Králové 1998; 43 : 127–132.
5. Skácel Z, Marel M, Vraštilová P et al. Histiocytóza z Langerhansových buněk. Přehled literatury a vlastní pozorování. Stud Pneumol Phthiseol 2000; 60 : 150–156.
6. Kinkor Z. Severe pulmonary involvement in Erdheim-Chester disease (case report). Cesk Patol 2001; 37 : 114–147.
7. Pacovská V, Bortlová A, Homolka J et al. Granulomatóza z Langerhansových buněk. Trendy Med 2002; 4 : 59–61.
8. Kodet R, Elleder M, Šmelhaus V et al. Disseminated histiocytosis X. Cesk Patol 1984; 20 : 19–26.
9. Drastík J. Histiocytosis X in otorhinolaryngology. Cesk Otolaryngol 1970; 19 : 49–55.
10. Fassmann A, Horáček J, Šindekla Z et al. Histocytosis X classified as Hand-Schueller-Christian disease in the oral cavity. Cesk Stomatol 1982; 82 : 32–37.
11. Hrubala D, Durovic E, Markovská N. Histiocytosis X. Prakt Zubn Lek 1984; 32 : 73–75.
12. Adam Z, Pour L, Krejčí M et al. Histiocytóza z Langerhansových buněk u osob dospělého věku nemoc s mnoha tvářemi. Zkušenosti jednoho pracoviště a přehled projevů nemoci. Vnitř Lék 2008; 54 : 1063–1081.
13. Anton M, Holoušova M, Řehůřek J et al. Histiocytoza X a dětská očnice. Čs Ophthal 1992; 48 : 176–180.
14. Bláha M. The hemophagocytic syndrome. Vnitř Lék 2003; 49 : 163–164.
15. Bunanská E, Stančoková T, Dluholucký S. Histiocytóza z Langerhansových buněk. Čes Slov Pediat 1998; 53 : 18–19.
16. Kodet R, Zitková M. Morphologic and roentgenologic pulmonary findings in disseminated histiocytosis X. Cesk Pediatr 1985; 40 : 634–638.
17. Kodetová D, Kodet R, Syrůček M et al. Sinus histiocytosis with massive lymphadenopathy – a disseminated form of the Rosai-Dorfman syndrome. Cesk Patol 1996; 32 : 53–59.
18. Mlyncek M, Uharcek P, Durcanský D. Vulvar Langerhans’ cell histiocytosis: a case report. Acta Obstet Gynecol Scand 2006; 85 : 753–755.
19. Mottl H, Ganevová M, Radvanská J et al. Treatment results of Langerhans cell histiocytosis with LSH II protocol. Cas Lek Cesk 2005; 144 : 753–755.
20. Mottl H, Koutecký J, Ganevová M. Strategie léčby histiocytózy z Langerhansových buněk u dětí. Čes Slov Pediat 1994; 49 : 81.
21. Mottl H, Mráček J Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530–533.
22. Mottl H, Mrácek J, Kabelka Z et al. Langerhans-cell histiocytosis in children. Cesk Pediatr 1992; 47 : 530–533.
23. Mottl H, Rob L, Stary J et al. Langerhans cell histiocytosis of vulva in adolescent. Int J Gynecol Cancer 2000; 17 : 520–524.
24. Pazdera J, Cerný O, Jirava E et al. Histiocytosis X of Abt-Letterer-Siwe type with primary manifestation in the oral cavity in a 15-months-old child. Cesk Stomatol 1974; 74 : 136–141.
25. Schwartz A, Zich P. Eosinophilic histiocytic granuloma of the vulva and cervix. Sb Lek 1989; 91 : 1–4.
26. Smilek P, Krejčová B, Čada K et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994; 43 : 263–265.
27. Ščudla V, Roček V, Dušek B et al. Multifokální eozinofilní granulom v dospělosti. Vnitř Lék 1987; 33 : 1078–1086.
28. Šímová B, Mališ J, Neuwirt J. Klinické projevy histiocytózy z Langerhansových buněk. Zdrav Nov ČR Lék Listy 2003; 52 : 18.
29. Tichy M Jr, Tichy M, Krč I et al. Multicentrická retikulohistiocytóza. Čes Slov Derm 1999; 74 : 168–171.
30. Toušovská K, Slavík Z. Histiocytóza z Langerhansových buněk v dětském věku. Lék Zprav Lék Fak Univ Karlovy Hr Králové 1997; 42 : 127–132.
31. Velková A. Changes in the periodontium in children. Cesk Stomatol 1975; 75 : 180–184.
32. Krajicek BJ, Ryu JH, Hartman TE et al. Abnormal fluorodeoxyglucose PET in pulmonary Langerhans cell histiocytosis. Chest 2009; 135 : 1542–1549.
33. Kawase T, Hamazaki M, Ogura M et al. CD56/NCAM-positive Langerhans cell sarcoma: a clinicopathologic study of 4 cases. Int J Hematol 2005; 81 : 323–329.
34. Lee JS, Ko GH, Kim HC et al. Langerhans cell sarcoma arising from Langerhans cell histiocytosis: a case report. J Korean Med Sci 2006; 21 : 577–580.
35. Willman CL, Busque L, Griffith BB et al. Langerhans’-cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med 1994; 331 : 154–160.
36. Yu RC, Chu C, Buluwela L et al. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994; 343 : 767–768.
37. Yousem SA, Colby TV, Chen YY et al. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol 2001; 25 : 630–636.
38. Geissmann F, Dieu-Nosjean MC, Dezutter C et al. Accumulation of immature Langerhans cells in human lymph nodes draining chronically inflamed skin. J Exp Med 2002; 196 : 417–430.
39. Geissmann F, Lepelletier Y, Fraitag S et al. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood 2001; 97 : 1241–1248.
40. Tazi A, Bonay M, Grandsaigne M et al. Surface phenotype of Langerhans cells and lymphocytes in granulomatous lesions from patients with pulmonary histiocytosis X. Am Rev Respir Dis 1993; 147 : 1531–1536.
41. Tazi A, Bouchonnet F, Grandsaigne M et al. Evidence that granulocyte macrophage-colony-stimulating factor regulates the distribution and differentiated state of dendritic cells/Langerhans cells in human lung and lung cancers. J Clin Invest 1993; 91 : 566–576.
42. Tazi A, Bonay M, Bergeron A et al. Role of granulocyte-macrophage colony stimulating factor (GM-CSF) in the pathogenesis of adult pulmonary histiocytosis X. Thorax 1996; 51 : 611–614.
43. Tazi A, Montcelly L, Bergeron A et al. Relapsing nodular lesions in the course of adult pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 1998; 157 : 2007–2010.
44. Tazi A, Moreau J, Bergeron A et al. Evidence that Langerhans cells in adult pulmonary Langerhans cell histiocytosis are mature dendritic cells: importance of the cytokine microenvironment. J Immunol 1999; 163 : 3511–3515.
45. Arrighi JF, Soulas C, Hauser C et al. TNF-a induces the generation of Langerin/(CD207)+ immature Langerhans-type dendritic cells from both CD14–CD1a and CD14+CD1a – precursors derived from CD34+ cord blood cells. Eur J Immunol 2003; 33 : 2053–2063.
46. Banchereau J, Briere F, Caux C et al. Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18 : 767–811.
47. Brabencova E, Tazi A, Lorenzato M et al. Langerhans cells in Langerhans cell granulomatosis are not actively proliferating cells. Am J Pathol 1998; 152 : 1143–1149.
48. Nagarjun Rao R, Moran CA, Suster S. Histiocytic disorders of the lung. Adv Anat Pathol 2010; 17 : 12–22.
49. Hance AJ, Basset F, Saumon G et al. Smoking and interstitial lung disease. The effect of cigarette smoking on the incidence of pulmonary histiocytosis X and sarcoidosis. Ann NY Acad Sci 1986; 465 : 643–656.
50. Banchereau J, Paczesny S, Blanco P et al. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann NY Acad Sci 2003; 987 : 180–187.
51. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell 2001; 106 : 255–258.
52. Valladeau J, Duvert-Frances V, Pin JJ et al. The monoclonal antibody DCGM4 recognizes langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur J Immunol 1999; 29 : 2695–2704.
53. Vassallo R, Jensen EA, Colby TV et al. The overlap between respiratory bronchiolitis and desquamative interstitial pneumonia in pulmonary Langerhans cell histiocytosis: high-resolution CT, histologic, and functional correlations. Chest 2003; 124 : 1199–1205.
54. Kambouchner M, Basset F, Marchal J et al. Three-dimensional characterization of pathologic lesions in pulmonary langerhans cell histiocytosis. Am J Respir Crit Care Med 2002; 166 : 1483–1490.
55. Watanabe R, Tatsumi K, Hashimoto S et al. Respiratory Failure Research Group of Japan. Clinico-epidemiological features of pulmonary histiocytosis X. Intern Med 2001; 40 : 998–1003.
56. Egeler RM, Neglia JP, Puccetti DM et al. Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer 1993; 71 : 865–873.
57. Neumann MP, Frizzera G. The coexistence of Langerhans’ cell granulomatosis and malignant lymphoma may take different forms: report of seven cases with a review of the literature. Hum Pathol 1986; 17 : 1060–1065.
58. Unger J, England D, Collins J. Miliary nodules, Hodgkin’s disease, and eosinophilic granuloma. J Thorac Imaging 1994; 9 : 71–73.
59. Gaensler E, Carrington C. Open biopsy for chronic diffuse infiltrative lung disease: clinical, roentgenographic, and physiological correlations in 502 patients. Ann Thorac Surg 1980; 30 : 411–426.
60. Colby TV, Lombard C. Histiocytosis X in the lung. Hum Pathol 1983; 14 : 847–856.
61. Thomeer M, Demedts M, Vandeurzen K. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg 2001; 56 : 163–172.
62. Kim CK, Park CB, Jin U et al. Pulmonary Langerhans’ cell histiocytosis presented with recurrent pneumothorax. Interact Cardiovasc Thorac Surg 2006; 5 : 512–513.
63. Krawczyk P, Czekajska-Chehab E, Kieszko R et al. Difficulties in the diagnosis of rare immunological diseases manifesting with cystic lung disease and spontaneous pneumothorax: case reports. Heart Lung 2004; 33 : 21–25.
64. Mendez JL, Nadrous HF, Vassallo R et al. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125 : 1028–1032.
65. Minghini A, Trogdon SD. Recurrent spontaneous pneumothorax in pulmonary histiocytosis X. Am Surg 1998; 64 : 1040–1042.
66. Knight RK. Haemoptysis in eosinophilic granuloma. Br J Dis Chest 1979; 73 : 181–186.
67. Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27 : 1272–1285.
68. Yağci B, Varan A, Cağlar M et al. Langerhans cell histiocytosis: retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol 2008; 25 : 399–408.
69. Lieberman PH, Jones CR, Steinman RM et al. Langerhans cell (eosinophilic) granulomatosis. A clinicopathologic study encompassing 50 years. Am J Surg Pathol 1996; 20 : 519–552.
70. Ryu JH, Colby TV, Hartman TE et al. Smoking-related interstitial lung diseases: a concise review. Eur Respir J 2001; 17 : 122–132.
71. ten Velde GP, Thunnissen FB, van Engelshoven JM et al. A solitary pulmonary nodule due to eosinophilic granuloma. Eur Respir J 1994; 7 : 1539–1540.
72. Brambilla E, Fontaine E, Pison CM et al. Pulmonary histiocytosis X with mediastinal lymph node involvement. Am Rev Respir Dis 1990; 142 : 1216–1218.
73. Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans’ cell histiocytosis. Thorax 2000; 55 : 405–416.
74. Vassallo R, Limper AH. Pulmonary Langerhans’ cell histiocytosis. Semin Respir Crit Care Med 2002; 23 : 93–101.
75. Vassallo R, Ryu JH, Colby TV et al. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000; 342 : 1969–1978.
76. Vassallo R, Ryu JH, Schroeder DR et al. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346 : 484–490.
77. Vassallo R, Ryu JH. Pulmonary Langerhans‘ cell histiocytosis. Clin Chest Med 2004; 25 : 561–571.
78. Sundar KM, Gosselin MV, Chung HL et al. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology, and clinical evolution of disease. Chest 2003; 123 : 1673–1683.
79. Lin MW, Chang YL, Lee YC et al. Pulmonary Langerhans cell histiocytosis. Lung 2009; 187 : 261–262.
80. Brauner MW, Grenier P, Mouelhi MM et al. Pulmonary histiocytosis X: evaluation with high-resolution CT. Radiology 1989; 172 : 255–258.
81. Brauner MW, Grenier P, Tijani K et al. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology 1997; 204 : 497–502.
82. Pipavath S, Godwin JD. Imaging of interstitial lung disease. Clin Chest Med. 2004; 25 : 455–465.
83. Moore AD, Godwin JD, Müller NL et al. Pulmonary histiocytosis X: comparison of radiographic and CT findings. Radiology 1989; 172 : 249–254.
84. Vrielynck S, Mamou-Mani T, Emond S et al. Diagnostic value of high-resolution CT in the evaluation of chronic infiltrative lung disease in children. AJR Am J Roentgenol 2008; 191 : 914–920.
85. Soler P, Bergeron A, Kambouchner M et al. Is high-resolution computed tomography a reliable tool to predict the histopathological activity of pulmonary Langerhans cell histiocytosis? Am J Respir Crit Care Med 2000; 162 : 264–270.
86. Aricò M, Egeler RM. Clinical aspects of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 247–258.
87. Aricò M, Haupt R, Russotto VS et al. Langerhans cell histiocytosis in two generations: a new family and review of the literature. Med Pediatr Oncol 2001; 36 : 314–316.
88. Friedman PJ, Liebow AA, Sokoloff J. Eosinophilic granuloma of lung. Clinical aspects of primary histiocytosis in the adult. Medicine (Baltimore) 1981; 60 : 385–396.
89. Crausman RS, Jennings CA, Tuder RM et al. Pulmonary histiocytosis X: pulmonary function and exercise pathophysiology. Am J Respir Crit Care Med 1996; 153 : 426–435.
90. Basset F, Corrin B, Spencer H et al. Pulmonary histiocytosis X. Am Rev Respir Dis 1978; 118 : 811–820.
91. Schönfeld N, Frank W, Wenig S et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993; 60 : 38–44.
92. Schönfeld N. Pulmonary Langerhans cell histiocytosis. Pneumologie 2003; 57 : 159–165.
93. Travis WD, Borok Z, Roum JH et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 1993; 17 : 971–986.
94. Wall CP, Gaensler EA, Carrington CB et al. Comparison of transbronchial and open biopsies in chronic infiltrative lung diseases. Am Rev Respir Dis 1981; 123 : 280–285.
95. Housini I, Tomashefski JF Jr, Cohen A et al. Transbronchial biopsy in patients with pulmonary eosinophilic granuloma. Comparison with findings on open lung biopsy. Arch Pathol Lab Med 1994; 118 : 523–530.
96. Mayer J, Skřičková J, Vorlíček J. Postižení plic u imunokompromitovaných nemocných. Diferenciální diagnostika a využití bronchoalveolární laváže. Brno: Institut pro další vzdělávání pracovníků ve zdravotnictví 1995.
97. Hance AJ. Pulmonary immune cells in health and disease: dendritic cells and Langerhans’ cells. Eur Respir J 1993; 6 : 1213–1220.
98. Soler P, Moreau A, Basset F et al. Cigarette smoking-induced changes in the number and differentiated state of pulmonary dendritic cells/Langerhans cells. Am Rev Respir Dis 1989; 139 : 1112–1117.
99. Casolaro MA, Bernaudin JF, Saltini C et al. Accumulation of Langerhans’ cells on the epithelial surface of the lower respiratory tract in normal subjects in association with cigarette smoking. Am Rev Respir Dis 1988; 137 : 406–411.
100. Auerswald U, Barth J, Magnussen H. Value of CD-1-positive cells in bronchoalveolar lavage fluid for the diagnosis of pulmonary histiocytosis X. Lung 1991; 169 : 305–309.
101. Chollet S, Soler P, Dournovo P et al. Diagnosis of pulmonary histiocytosis X by immunodetection of Langerhans cells in bronchoalveolar lavage fluid. Am J Pathol 1984; 115 : 225–232.
102. Colasante A, Poletti V, Rosini S et al. Langerhans cells in Langerhans cell histiocytosis and peripheral adenocarcinomas of the lung. Am Rev Respir Dis 1993; 148 : 752–759.
103. Takizawa Y, Taniuchi N, Ghazizadeh M et al. Bronchoalveolar lavage fluid analysis provides diagnostic information on pulmonary Langerhans cell histiocytosis. J Nippon Med Sch 2009; 76 : 84–92.
104. Tötsch M, Guzman J, Theegarten D et al. Bronchoalveolar lavage. Pathologe 2007; 28 : 346–353.
105. Zeppa P, Cozzolino I, Russo M et al. Pulmonary langerhans cell histiocytosis (histiocytosis X) on bronchoalveolar lavage: a report of 2 cases. Acta Cytol 2007; 51 : 480–482.
106. Krenács L, Tiszalvicz L, Krenács T et al. Immunohistochemical detection of CD1A antigen in formalin-fixed and paraffin-embedded tissue sections with monoclonal antibody 010. J Pathol 1993; 171 : 99–104.
107. Wang CW, Colby TV. Histiocytic lesions and proliferations in the lung. Semin Diagn Pathol 2007; 24 : 162–182.
108. Emile JF, Wechsler J, Brousse N et al. Langerhans’ cell histiocytosis. Definitive diagnosis with the use of monoclonal antibody O10 on routinely paraffin-embedded samples. Am J Surg Pathol 1995; 19 : 636–641.
109. Howarth DM, Gilchrist GS, Mullan BP et al. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer 1999; 85 : 2278–2290.
110. Johnson S. Rare diseases. 1. Lymphangioleiomyomatosis: clinical features, management and basic mechanisms. Thorax 1999; 54 : 254–264.
111. Muzzi L, Pini Prato GP, Ficarrat G. Langerhans’ cell histiocytosis diagnosed through periodontal lesions: a case report. J Periodontol 2002; 73 : 1528–1533.
112. Nagai S. Pulmonary Langerhans’ cell histiocytosis (LCH): epidemiology and clinical courses. Intern Med 2001; 40 : 981–982.
113. Ouyang DL, Roberts BK, Gibbs IC et al. Isolated Langerhans cell histiocytosis in an adult with central diabetes insipidus: case report and review of literature. Endocr Pract 2006; 12 : 660–663.
114. Powers MA, Askin FB, Cresson DH. Pulmonary eosinophilic granuloma. 25-year follow-up. Am Rev Respir Dis 1984; 129 : 503–507.
115. Unel SY, Ozaslan H, Solmaz D et al. Reversible pulmonary Langerhans’ cell histiocytosis. JBR-BTR 2009; 92 : 172–173.
116. Valliani L, Kanwar VS, Schwartz A. Isolated pulmonary Langerhans cell histiocytosis with recurrent bilateral pneumothoraces treated with chemotherapy and chemical pleurodesis. Pediatr Blood Cancer 2009; 53 : 128–129.
117. Marten K. Smoking-related interstitial lung diseases. Rofo 2007; 179 : 268–275.
118. Mogulkoc N, Veral A, Bishop PW et al. Pulmonary Langerhans’ cell histiocytosis: radiologic resolution following smoking cessation. Chest 1999; 115 : 1452–1455.
119. Negrin-Dastis S, Butenda D, Dorzee J e al. Complete disappearance of lung abnormalities on high-resolution computed tomography: a case of histiocytosis X. Can Respir J 2007; 14 : 235–237.
120. Tomashefski JF, Khiyami A, Kleinerman J. Neoplasms associated with pulmonary eosinophilic granuloma. Arch Pathol Lab Med 1991; 115 : 499–506.
121. Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet 1987; 1 : 208–209.
122. Pappas CA, Rheinlander HF, Stadecker MJ. Pleural effusion as a complication of solitary eosinophilic granuloma of the rib. Hum Pathol 1980; 11 : 675–677.
123. Delobbe A, Durieu J, Duhamel A et al. Groupe d’Etude en Pathologie Interstitielle de la Société de Pathologie Thoracique du Nord. Determinants of survival in pulmonary Langerhans’ cell granulomatosis (histiocytosis X). Eur Respir J 1996; 9 : 2002–2006.
124. Fartoukh M, Humbert M, Capron F et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med 2000; 161 : 216–223.
125. Favara BE, Feller AC, Pauli M et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997; 29 : 157–166.
126. Hamada K, Teramoto S, Narita N et al. Pulmonary veno-occlusive disease in pulmonary Langerhans’ cell granulomatosis. Eur Respir J 2000; 15 : 421–423.
127. Lombard CM, Medeiros LJ, Colby TV. Pulmonary histiocytosis X and carcinoma. Arch Pathol Lab Med 1987; 111 : 339–341.
128. Sadoun D, Vaylet F, Valeyre D et al. Bronchogenic carcinoma in patients with pulmonary histiocytosis X. Chest 1992; 101 : 1610–1613.
129. Zeid NA, Muller HK. Tobacco smoke induced lung granulomas and tumours: association with pulmonary Langerhans cells. Pathology 1995; 27 : 247–254.
130. Von Essen S, West W, Sitorius M et al. Complete resolution of roentgenographic changes in a patient with pulmonary histiocytosis X. Chest 1990; 98 : 765–767.
131. Malpas JS. Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Am 1998; 12 : 259–268.
132. Boehler A. Lung transplantation for cystic lung diseases: lymphangioleiomyomatosis, histiocytosis X, and sarcoidosis. Semin Respir Crit Care Med 2001; 22 : 509–515.
133. Sulica R, Teirstein A, Padilla ML. Lung transplantation in interstitial lung disease. Curr Opin Pulm Med 2001; 7 : 314–322.
134. Etienne B, Bertocchi M, Gamondes JP et al. Relapsing pulmonary Langerhans cell histiocytosis after lung transplantation. Am J Respir Crit Care Med 1998; 157 : 288–291.
135. Gabbay E, Dark JH, Ashcroft T et al. Recurrence of Langerhans’ cell granulomatosis following lung transplantation. Thorax 1998; 53 : 326–327.
136. Habib SB, Congleton J, Carr D et al. Recurrence of recipient Langerhans’ cell histiocytosis following bilateral lung transplantation. Thorax 1998; 53 : 323–325.
137. Kasmani R, Narwal-Chadha R, Naveed S et al. Isolated pulmonary Langerhans cell histiocytosis. QJM 2009; 102 : 741–742.
138. Konno S, Hizawa N, Betsuyaku T et al. Adult Langerhans cell histiocytosis with independently relapsing lung and liver lesions that was successfully treated with etoposide. Intern Med 2007; 46 : 1231–1235.
139. Koschel D, Müller-Wening D. Pulmonary Langerhans-cell histiocytosis. Med Klin (Munich) 2003; 98 : 461–463.
140. Lacronique J, Roth C, Battesti JP et al. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Torax 1982; 37 : 104–109.
141. Saven A, Burian C. Cladribine activity in adult Langerhans-cell histiocytosis. Blood 1999; 93 : 4125–4130.
142. Pardanani A, Phyliky RL, Li CY et al. 2-Chlorodeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc 2003; 78 : 301–306.
143. Goh NS, McDonald CE, MacGregor DP et al. Successful treatment of Langerhans cell histiocytosis with 2-chlorodeoxyadenosine. Respirology 2003; 8 : 91–94.
144. Lazor R, Etienne-Mastroianni B, Khouatra C et al. Progressive diffuse pulmonary Langerhans cell histiocytosis improved by cladribine chemotherapy. Thorax 2009; 64 : 274–275.
145. Weitzman S, Braier J, Donadieu J et al. 2’-chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). Results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 2009; 53 : 1271–1276.
146. von Stebut E, Schadmand-Fischer S, Bräuninger W et al. Successful treatment of adult multisystemic Langerhans cell histiocytosis with psoralen-UV-A, prednisolone, mercaptopurine and vinblastine. Arch Dermatol 2008; 144 : 649–653.
147. Henter JI, Karlén J, Calming U et al. Successful treatment of Langerhans’-cell histiocytosis with etanercept. N Engl J Med 2001; 345 : 1577–1578.
148. McClain KL, Kozinetz CA. A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 2007; 48 : 44–49.
149. Rodríguez HN, García I, Alba A et al. Infliximab-induced reactivated Langerhan’s cell histiocytosis in a patient with ulcerative colitis. Inflamm Bowel Dis 2009; 15 : 1286–1287.
150. Calming U, Jacobsson H, Henter JI. Detection of Langerhans cell histiocytosis lesions with somatostatin analogue scintigraphy – a preliminary report. Med Pediatr Oncol 2000; 35 : 462–467.
151. Weinmann P, Crestani B, Tazi A et al. 111In-pentetreotide scintigraphy in patients with Langerhans’ cell histiocytosis. J Nucl Med 2000; 41 : 1808–1812.
152. Lastoria S, Montella L, Catalano L et al. Functional imaging of Langerhans cell histiocytosis by 111In-DTPA-D-Phe(1)-octreotide scintigraphy. Cancer 2002; 94 : 633–640.
153. Weiss SE, O’Connor L, Welsh JS. Refinement of radiation therapy based on PET data in an adult with Langerhans cell histiocytosis of soft tissues. Clin Adv Hematol Oncol 2006; 4 : 290–292.
154. Steiner M, Prayer D, Asenbaum S et al. Modern imaging methods for the assessment of Langerhans’ cell histiocytosis-associated neurodegenerative syndrome: case report. J Child Neurol 2005; 20 : 253–257.
155. Schmidt GP, Reiser MF, Baur-Melnyk A. Whole-body imaging of bone marrow. Semin Musculoskelet Radiol 2009; 13 : 120–133.
156. Derenzini E, Fina MP, Stefoni V et al. MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis: experience on seven patients. Ann Oncol 2010; 21 : 1173–1178.
157. Phillips M, Allen C, Gerson P et al. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer 2009; 52 : 97–101.
158. Ribeiro MJ, Idbaih A, Thomas C et al. 18F-FDG PET in neurodegenerative Langerhans cell histiocytosis: results and potential interest for an early diagnosis of the disease. J Neurol 2008; 255 : 575–580.
159. Arnaud L, Malek Z, Archambaud F et al. 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum 2009; 60 : 3128–3138.
160. Tan IB, Padhy AK, Thng CH et al. Intensely hypermetabolic extra-axial brainstem tumor in Erdheim-Chester disease. Clin Nucl Med 2009; 34 : 604–607.
161. Takahashi T, Yoshimoto M, Kondoh N. Spontaneously regressed Langerhans cell histiocytosis of lymph nodes in an elderly patient. Intern Med 2007; 46 : 1757–1760.
162. Kaste SC, Rodriguez-Galindo C, McCarville ME et al. PET-CT in pediatric Langerhans cell histiocytosis. Pediatr Radiol 2007; 37 : 615–622.
163. Binkovitz LA, Olshefski RS, Adler BH. Coincidence FDG-PET in the evaluation of Langerhans’ cell histiocytosis: preliminary findings. Pediatr Radiol 2003; 33 : 598–602.
164. Blum R, Seymour JF, Hicks RJ. Role of FDG-positron emission tomography scanning in the management of histiocytosis. Leukem Lymphoma 2002; 43 : 2155–2157.
165. Calming U, Bemstrand C, Mosskin M et al. Brain 18-FDG PET scan in central nervous system langerhans cell histiocytosis. J Pediatr 2002; 141 : 435–440.
166. Aoki J, Watanabe H, Shinozaki T et al. FDG PET of primary benign and malignant bone tumors: standardized uptake value in 52 lesions. Radiology 2001; 219 : 774–777.
167. Giovanella L, Ceriani L, Crippa S et al. Imaging in endocrinology: Langerhans cell histiocytosis of the thyroid gland detected by 18FDG-PET/CT. J Clin Endocrinol Metab 2007; 92 : 2866–2867.
168. Büchler T, Cervinek L, Belohlavek O et al. Langerhans cell histiocytosis with central nervous system involvement: follow-up by FDG-PET during treatment with cladribine. Pediatr Blood Cancer 2005; 44 : 286–288.
169. Feurestein IM, Archer A, Pluda JM et al. Thin-walled cavities, cysts, and pneumothorax in Pneumocystis carinii pneumonia: further observations with histopathologic correlation. Radiology 1990; 174 : 697–702.
170. Khoor A, Myers JL, Tazelaar HD et al. Pulmonary Langerhans cell histiocytosis presenting as a solitary nodule. Mayo Clin Proc 2001; 76 : 209–211.
171. Fichter J, Doberauer C, Seegenschmiedt H. Langerhans cell histiocytosis in adults: An interdisciplinary challenge. Dtsch Arztebl 2007; 104: A2347–A2353.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2010 Číslo Supplementum 2
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících