
Histiocytóza z Langerhansových buněkz pohledu patologa
Langerhans cell histiocytosis: a pathologist view
Langerhans cell histiocytosis is a clinico - pathological entity with a wide spectrum of clinical and morphological findings. The disease was defined on the basis of recognition of three entities in medical history – Hand - Schüller - Christian disease, Letterer - Siwe disease and eosinophilic granuloma of bone. Later on in the past, these diseases were linked under a term histiocytosis X. With identification of a common cell of origin, the Langerhans cell, a name “Langerhans cell histiocytosis” (LCH) has been accepted. This review summarizes more than a hundred-year evolution of views on the disease. Langerhans cells are identified with the aid of histopathological investigations and an ultrastructural demonstration of specific membranous intracytoplasmic structures – Birbeck granules. At present the diagnosis of LCH utilizes immunohistochemical investigations to demonstrate positivity of S-100 protein, CD1a and langerin (CD207) in Langerhans cells. Characteristics of these molecules are briefly summarized. Further supportive laboratory methods may be used to demonstrate expression of proteins of the cell cycle, namely Ki - 67. The evaluation of the proliferation activity might support the estimation of a potential of the disease to progress or disseminate, especially in cases diagnosed at early stages or in situations they involve a single organ or tissue, and the progression might evolve secondarily. The histopathological differential diagnosis should separate LCH from reactive non-neoplastic Langerhans cell proliferations, histocytic diseases and a number of neoplastic diseases which are briefly reviewed.
Key words:
Langerhans cell histiocytosis – classification – histopathological and immunophenotypical findings – differential diagnosis
Autoři:
R. Kodet; M. Mrhalová
Působiště autorů:
Ústav patologie a molekulární medicíny 2. lékařské fakulty UK a FN Motol Praha, přednosta prof. MU Dr. Roman Kodet, CSc.
Vyšlo v časopise:
Vnitř Lék 2010; 56(Supplementum 2): 27-38
Kategorie:
Histiocytóza z Langerhansových buněk a některá další vzácná hematologická onemocnění
Souhrn
Histiocytóza z Langerhansových buněk je klinicko-patologickou jednotkou s variabilními klinickými a morfologickými nálezy. Její definice byla formulována na základě postupného rozpoznání 3 jednotek – chorob Hand - Schüller - Christianovy, Letterer - Siweovy a eozinofilního granulomu kostí. Později byly tyto jednotky sloučeny pod názvem histiocytóza X. S identifikací společného jmenovatele onemocnění – Langerhansových buněk – byl zaveden název histiocytóza z Langerhansových buněk (LCH). V přehledu shrnujeme více než 100 let vývoje poznatků o LCH. Langerhansovy buňky lze rozpoznat na základě histopatologického vyšetření a ultrastrukturálním průkazem specifických membránových intracytoplazmatických struktur – Birbeckových granul. V současnosti se pro podporu diagnózy LCH využívá imunohistochemické vyšetření, které u Langerhansových buněk prokazuje expresi S-100 proteinu, molekuly CD1a a langerinu (CD207). Tyto molekuly jsou v krátkém přehledu charakterizovány. Využitelnost dalších diagnosticky podpůrných metod zahrnuje aplikaci markerů buněčného cyklu, zejména molekuly Ki - 67. Posouzení proliferační aktivity onemocnění by mohlo mít význam v odhadu dalšího vývoje nemoci, zejména zachycené v časném stadiu vývoje nebo při monoorgánové formě, u které by mohla nastat progrese a generalizace druhotně. Histopatologická diferenciální diagnóza LCH musí odlišit tuto chorobu od nenádorových reaktivních změn Langerhansových buněk, histiocytárních chorob i od četných dalších nádorových procesů, jejichž spektrum uvádíme v přehledu.
Klíčová slova:
histiocytóza z Langerhansových buněk – klasifikace – histopatologické a imunofenotypické nálezy – diferenciální diagnóza
Úvod
Objasnění a vyčlenění klinicko-patologické jednotky a názvosloví
Histiocytóza z Langerhansových buněk (LCH) je skupinou onemocnění vycházejících z dendritických Langerhansových buněk. Projevy a biologické chování jsou mimořádně variabilní. Onemocnění může být generalizované s rychlým a smrtícím průběhem nebo na druhém konci spektra projevů může být lokalizované a chronické. Postiženy bývají většinou děti, avšak onemocnění se vyskytuje i u dospělých [1]. Míra agresivity a generalizace choroby mají k věku nemocných přímý vztah. Obecně platí, čím mladší jedinec, tím rizikovější je průběh nemoci. Generalizované formy s rychlým průběhem postihují převážně děti v kojeneckém a batolecím věku, zatímco lokalizované formy jsou častější u starších dětí a u dospělých nemocných. LCH je pevně zakotvena ve WHO klasifikaci nádorů hemopoetického a lymfoidního systému. Kromě vlastní „typické LCH“ je zde samostatně vyčleněna jednotka „sarkom z Langerhansových buněk“ [2]. Jde o mimořádně vzácné onemocnění i ve vztahu k frekvenci výskytu vzácné samotné LCH a krátce o něm pojednáme na konci této statě. Přehledný článek o histiocytózách, včetně LCH klasifikace a terapie, podává Adam et al v supplementu 1 tohoto časopisu z roku 2009 [3].
Názory na LCH se rodily v první polovině minulého století, resp. s prvními popisy dr. Alfreda Handa od konce 19. století. Nezávisle na tom byly publikovány další případy, a to v letech 1915 – 1916 (Arthur Schüller) a 1920 (Henry Asbury Christian). Ve 20. letech 20. století byly tyto případy referovány jako onemocnění Christianovo, pak Schüllerovo a Christianovo a nakonec bylo do názvu včleněno také jméno A. Handa jako autora do té doby prvního známého popisu nemoci. V roztříštěném názvosloví se tedy ustálilo označení buď Schüller-Christianova, nebo Hand-Schüller-Christianova choroba [4]. Uvažuje se však, že vůbec první popis choroby uvedl Thomas Smith v roce 1865 [4]. Onemocnění bylo pomalu progresivní s iniciálně popsanou trias zahrnující osteolytické kostní defekty, exoftalmus a diabetes insipidus. Druhou, nezávisle popsanou skupinou bylo agresivní a rychle probíhající letální onemocnění Letterer-Siweovo (L-S) – název zvolili Arthur F. Abt a Edward J. Denenholz v roce 1936 [5]. Nejpozději, v letech 1940 – 1942, bylo identifikováno také lokalizované postižení kostí – eozinofilní granulom. Protože postižení kostí tímto onemocněním vyvolávalo v té době velkou pozornost, vznikla kolem rozpoznání charakteru osteolytických kostních lézí histiocytárního charakteru malá epidemie publikací [6 – 8]. Již ve 40. letech minulého století se však začalo uvažovat, že uvedené choroby mají společného jmenovatele [9 – 12] a někteří autoři poukazovali na překryvy těchto jednotek a na skutečnost, že některé případy nelze jednoduše zařadit do jednotlivých kategorií. Vznikaly první návrhy na sjednocení uvedených jednotek pod společného jmenovatele. První takový pokus provedli Dennis a Rosahn v roce 1951, kteří navrhli označení „primární retikuloendoteliální granulom“ [13]. Avšak teprve v roce 1953 sjednotil pohled na onemocnění Louis Lichtenstein ve své práci publikované v časopisu Archives of Patology [14]. Lichtenstein koncipoval základní principy morfologických nálezů a zejména klinického průběhu nemoci. Původně izolované jednotky, které po stránce průběhu označil jako akutní nebo subakutní diseminované onemocnění (syndrom Letterer-Siwe – L-S) a subchronicky nebo chronicky probíhající chorobu (syndrom Schüller-Christian – S-C), sloučil jako formy diseminované histiocytózy. K tomu přiřadil eozinofilní granulom kosti jako lokalizovanou formu téhož onemocnění, bez viscerálního postižení. Pro uvedené syndromy zvolil zastřešující název – histiocytosis X. Písmeno X uvedl Lichtenstein proto, že mělo krátce a výstižně vyjadřovat značnou variabilitu projevů nemoci a mělo být stimulem k pátrání po její etiologii, která nebyla jasná. Zároveň doložil, že mezi uvedenými jednotkami jsou přechodné formy a že akutní/ subakutní diseminovaná forma (L-S) může přejít v chronickou (S-C) a naopak, subchronická/ chronická forma onemocnění může akcelerovat do podoby s akutní. V té době se onemocnění sice považovalo za histiocytární, jak již název napovídal, ale teprve ultrastrukturální pozorování (elektronová mikroskopie) odhalilo, že se jedná o specializovanou formu histiocytů, v dnešní době správněji buněk zařazených do skupiny buněk dendritických. Tyto buňky byly označeny jako buňky Langerhansovy. Dopomohla k tomu identifikace svérázných diskoidních intracytoplazmatických membranózních struktur v Langerhansových buňkách epidermis, odlišných od melanozomů v melanocytech [15]. Jak se ukázalo, tyto struktury, později nazvané jako granula X nebo Birbeckova granula, slouží jako buněčně specifický ultrastrukturální znak pro skupinu dendritických buněk označených jako Langerhansovy buňky. V krátké době byla Birbeckova granula prokázána i u histiocytózy X [16,17] a tento nález vedl k ucelené představě o původu buněk v rámci všech projevů histiocytózy X [18]. Stále však existovaly dohady, jak LCH vzniká. Původní práce chorobu S-C řadily mezi střádavé metabolické choroby, jakými jsou choroba Nieman-Pickova nebo Gaucherova. Tyto názory byly podrobeny kritice a vyvráceny v řadě pozdějších prací [11]. Eozinofilní granulom kosti byl pod názvem „solitární granulom kosti“ považován za benigní granulomatózní proces o neznámé příčině a nebyl vyloučen potraumatický [6] nebo virový [7] původ. O LCH se ještě v 80. letech minulého století spekulovalo jako o nenádorové reaktivní chorobě schopné spontánně regredovat [19]. V současnosti je prokázáno, že LCH je ve většině případů klonálním onemocněním [20,21]. Výjimku tvoří separátně klasifikované plicní histiocytózy z Langerhansových buněk u dospělých, které vznikají polyklonální proliferací Langerhansových buněk a vývoj tohoto onemocnění je přímo spjat s kuřáctvím [22]. I když průkaz klonality proliferujících buněk není považován za dostatečný důkaz nádorové povahy, je zřejmé, že u individuálních nemocných jde o poruchu jedné (klonální) populace Langerhansových buněk a onemocnění je nádoru přinejmenším blízké, obdobně jako některá monoklonální lymfoproliferativní onemocnění. Dosud nebyl identifikován žádný molekulárně specifický znak LCH, ale pro nádorovou – autonomní povahu svědčí některé poruchy klíčových regulačních proteinů buněčného cyklu [23], cytogenetické změny [24] a také průkaz ztráty heterozygozity některých chromozomálních úseků [25].
Po rozpoznání charakteru buněk v infiltrátech onemocnění histiocytózou X jako buněk Langerhansových byl doporučen nový název, a to histiocytóza z Langerhansových buněk, LCH [26]. Tento název se ujal a je běžně používán, včetně závazné WHO klasifikace nádorů krve a krvetvorných orgánů [2].
Histopatologický obraz LCH
Langerhansovy buňky jsou v histopatologickém vyšetření charakteristické a liší se od tkáňových histiocytů – makrofágů jak na úrovni světelné mikroskopie při běžném barvení a v expresi charakteristických proteinů, tak na úrovni ultrastrukturální.
Ve světelném mikroskopu jsou Langerhansovy buňky střední velikosti (obvykle kolem 12 – 15 µm), mají polygonální nebo mírně nepravidelně okrouhlý tvar a zřetelné buněčné hranice (obr. 1). Cytoplazma je v přehledném histologickém barvení hematoxylinem a eozinem homogenní, světle eozinofilní. Na rozdíl od epidermálních Langerhansových buněk netvoří v lézích LCH tyto buňky dendritické výběžky. Cytoplazma neobsahuje fagocytovaný materiál, fagocytované buňky ani pigmenty (např. hemosiderin nebo antrakotický pigment), jak tomu je u tkáňových makrofágů. Jádro Langerhansových buněk je typicky nepravidelně oválné, ledvinovité s charakteristickými invaginacemi jaderného povrchu (obr. 1). Někdy jsou na jádru patrné četné drobné indentace, připodobňované k mozkovým závitům a brázdám, tzv. cerebriformní charakter jader. Jadérka jsou drobná, nenápadná. Mitotická aktivita kolísá případ od případu, avšak u typických forem je nízká. Někdy ji nelze identifikovat vůbec. Infiltráty Langerhansových buněk při LCH mohou být monomorfní, ale často vídáme variabilní příměs velkých vícejaderných – mnohojaderných Langerhansových buněk. Není jisté, zda tyto mnohojaderné buňky vznikají fúzí mononukleárních forem nebo spíše dělením jader samotných. U kostních lézí a v lymfatických uzlinách jsou též často přítomny mnohojaderné buňky, v kostech charakteru osteoklastů, v uzlinách tzv. mnohojaderné buňky typu z cizích těles. V kostních ložiscích LCH zmnožené a metabolicky aktivní osteoklasty pravděpodobně zodpovídají za osteolytický charakter infiltrátů [27]. Infiltráty lézí LCH doplňuje příměs leukocytů, zejména T-lymfocytů a eozinofilních granulocytů (obr. 2). Eozinofilní leukocyty mohou místy až zakrývat podstatu infiltrátu z Langerhansových buněk nebo jsou tak hojné a rozpadají se, že se vytvářejí eozinofilní abscesy. Z rozpadlých granulocytů se někdy tvoří Charcot-Leydenovy krystaly. U perzistujících lézí klesá v infiltrátech podíl Langerhansových buněk s klasickým morfologickým obrazem a objevují se buňky pěnitého charakteru. Obraz pak silně připomíná pseudoxantomy při chronickém hnisání, např. ve stěně pyogenní membrány. V takových případech je rozpoznání povahy infiltrátu bez navazujících specializovaných vyšetření nebo bez přihlédnutí k anamnéze nemocného problematické. Na rozdíl od pěnitých histiocytů zánětlivého původu však bývají infiltráty steatotických buněk vysloveně masivní a tumoriformní, žluté barvy, zejména při postižení kostí a thymu, někdy i jiných orgánů. V perzistujících infiltrátech také přibývá stromálních buněk – myofibroblastů a extracelulární matrix, což odpovídá jizvení.


V rámci komplexu LCH jsou mikroskopické projevy variabilní. Při orgánové manifestaci nacházíme nejčastěji monomorfní uspořádání Langerhansových buněk (obr. 1), někdy s příměsí obrovských mnohojaderných buněk podobných osteoklastům původu tkáňových makrofágů. Při kostní formě LCH, monoostotické nebo polyostotické, tzv. eozinofilním granulomu kostí, je typická již zmíněná příměs eozinofilních leukocytů, která může ložiskově až početně převažovat nad Langerhansovými buňkami. Eozinofilní leukocyty se však v různé míře objevují i u orgánových manifestací LCH. Kostní ložiska jsou makroskopicky zřetelně osteolytická, tvoří souvislé masy měkké, šedorůžové až nažloutlé barvy. V okolí může být přítomna reaktivní osifikující periostitida. U starších lézí přibývá zmíněných pěnitých buněk a onemocnění může být mylně považováno za chronickou nespecifickou hnisavou osteomyelitidu. Poněkud odlišný je mikroskopický obraz v kožních lézích. V kůži jsou Langerhansovy buňky při LCH lokalizované v horní dermis a bývají zde hojnější T-lymfocyty. Langerhansovy buňky projevují výrazný epidermotropizmus, tj. pronikají přes bazální membránu mezi keratinocyty epidermis a při shlukování tvoří charakteristické vezikulární formace tvořené tekutinou s Langerhansovými buňkami a T-lymfocyty (obr. 3). U akutně probíhajících forem LCH s multiorgánovým postižením (dříve nemoc Letterer-Siwe) na tyto nálezy nasedají projevy hemoragické diatézy – hemoragie s přítomností erytrocytů v dermis a epidermis. Makroskopicky pak papulovesikulární projevy komplikuje hemoragický charakter [28]. Při mírnějším průběhu se kožní změny podobají nálezům u sebor-rhoické dermatitidy. Nejčastěji je postižena kůže vlasaté části hlavy, kde je také nejvíce zastoupena přirozená populace Langerhansových buněk. Infiltráty bývají také časté na trupu a případně na proximálních částech končetin. V rámci variability LCH může být postižení kůže součástí celkového onemocnění (u dětí), u starších nemocných může být kožní manifestace monoorgánová a jako jediná známka LCH (tzv. eozinofilní granulom kůže). Sami jsme pozorovali izolované kožní onemocnění u 4 nemocných ve věku 17, 27, 66 a 73 let (3 muži, 1 žena). U pacientky převažoval infiltrát v oblasti kůže přední hrudní stěny a v mikroskopické diferenciální diagnóze bylo nutné odlišit mycosis fungoides. Teprve pečlivé vyšetření jednotlivých buněk infiltrátu odhalilo přítomnost četných Langerhansových buněk v dermis i v epidermis. Netvořily se tzv. Pautrierovy mikroabscesy, ale typické vezikuly s četnými Langerhansovými buňkami. V lymfatických uzlinách je zpočátku patrná intrasinusoidální propagace Langerhansových buněk (obr. 4), později infiltráty přestupují do parakortexu a splývají v souvislé masy. Lymfatické uzliny mohou být postiženy v rámci systémového multiorgánového onemocnění, méně často jako vedlejší nález ve spádových uzlinách při drenáži z kostních ložisek. U starších nemocných bývají lymfatické uzliny postiženy samostatně [29]. Ve slezině je při LCH postižena převážně červená pulpa. V játrech bývají infiltráty vázány na intrahepatické žlučové cesty a při nepříznivém vývoji onemocnění vzniká v chronické fázi obraz sklerózující cholangitidy [30]. K hepatosplenomegalii u diseminovaných forem přispívají také kompaktnější tumoriformní infiltráty (obr. 5). Kromě uvedených orgánů jsou při systémové LCH postiženy plíce, thymus a někdy také centrální nervový systém. Formy a intenzita infiltrátů jsou velmi proměnlivé a liší se případ od případu. Souhrn morfologických nálezů u LCH je uveden v tab. 1.




Při regresi LCH po terapii Langerhansovy buňky, případně další buňky jako součást nenádorové zánětlivé příměsi někdy vymizí – např. z kůže. Jindy je regrese provázena jizvením – viz sklerózující cholangitida při jaterním postižení. Jizvení jsme pozorovali také v plicích s rozvojem tzv. voštinovité plíce a klinickými projevy respiračního selhání [31].
Kromě základního histopatologického vyšetření je důležitým diagnostickým znakem identifikace Langerhansových buněk na úrovni ultrastrukturální – průkaz tzv. Birbeckových granul v cytoplazmě. Birbeckova granula jsou membranózní pentalamelární struktury připomínající svým uspořádáním vzhled zipu (obr. 6). Sám Michael S. C. Birbeck ve svém popisu těchto struktur hovoří o granulech, ale postřehl, že mají spíše diskoidní tvar. V některých rovinách ultratenkého řezu se zachytí jejich dilatované úseky a pak granula tvarem připomínají tenisovou raketu. Přestože se prokážou prakticky ve všech případech LCH, procento Langerhansových buněk, u kterých lze granula prokázat, silně kolísá. Podle našich zkušeností byla v některých případech LCH Birbeckova granula velmi hojná, jindy je bylo třeba zdlouhavě hledat. V jedné detailnější studii se záchyt Birbeckových granul u jednotlivých případů pohyboval od 2 % do 79 % Langerhansových buněk [32]. To podle našich zkušeností odpovídá i heterogenní pozitivitě v imunohistochemickém průkazu langerinu, o kterém je pojednáno níže.

Z enzymů, které jsou typické pro Langerhansovy buňky, lze v diagnostice využít průkazu kyselé a-D-mannosidázy [33], avšak pro technickou náročnost se toto vyšetření v praxi neuplatnilo a nahrazuje jej průkaz exprese níže uvedených proteinů imunohistochemickými metodami.
Zařazení Langerhansových buněk a jejich imunofenotypické markery používané v diagnostické praxi
Langerhansovy buňky jsou jednou z forem buněk skupiny tzv. buněk dendritických. Jejich funkcí je prezentace antigenu T - a NK-lymfocytům. Dendritické buňky se rozdělují do 2 hlavních skupin – myeloidní a plazmacytoidní. Epidermální dendritické buňky (podle různých pramenů zastoupeny v 1 – 8 % všech epidermálních buněk) odpovídají v nezánětlivé kůži klasickým Langerhansovým buňkám. Jsou roztroušené mezi bazálními a suprabazálními keratinocyty. Mají zřetelně výběžkatou cytoplazmu, s výběžky vsunutými mezi jednotlivé keratinocyty (z toho důvodu byly svým objevitelem, P. Langerhansem, považovány za neurony – 1868). Leží při bazální části epidermálních buněk.
Langerhansovy buňky jsou derivované z monocytů. V epidermis jsou v „nezralém“ stavu, mají malý migrační potenciál a exprimují kostimulační molekuly v nízkých hladinách. Pro diferenciaci Langerhansových buněk je třeba transformující růstový faktor β (TGF-β) [34] a další molekuly. Dendritické buňky lokalizované v epidermis hrají klíčovou roli v rozpoznání antigenů a iniciují vrozenou i adaptivní odezvu. V normální kůži jsou přítomné 2 hlavní populace dendritických buněk – epidermální Langerhansovy buňky a dermální (nebo intersticiální) dendritické buňky. Langerhansovy buňky pocházejí z progenitorů v kostní dřeni. Proces diferenciace a migrace Langerhansových buněk do epidermis není zatím přesně prostudován. Podle jedné teorie se prekurzory Langerhansových buněk dostávají z periferní krve přes dermis do epidermis (prekurzory v kostní dřeni exprimují „myeloid cutaneous lymphocyte associated antigen“ – CLA). Jiné studie naznačují, že prekurzory Langerhansových buněk za normálních, nezánětlivých podmínek sídlí v dermis. Tyto prekurzory koexprimují langerin a molekulu CD14. Finální migrace těchto buněk do suprabazální vrstvy epidermis je pravděpodobně řízena impulzy z keratinocytů. Finální diferenciace prekurzorů Langerhansových buněk závisí na cytokinovém prostředí v epidermis. Langerhansovy buňky zpracují antigen. Poté migrují do lymfatických cév a s lymfatickou drenáží do parakortexu lymfatických uzlin. Během této migrace ztrácejí schopnost internalizovat další antigeny a naopak získávají schopnosti prezentovat antigen naivním T-lymfocytům (tzv. maturace) [35].
V normálních tkáních a v rámci LCH netvoří Langerhansovy buňky cytoplazmatické výběžky a neudržují mezi sebou žádné pevné kontakty. Vytvářejí se difuzní splývající infiltráty relativně uniformního charakteru.
Na úrovni exprese proteinů jsou Langerhansovy buňky charakterizované řadou molekul, avšak z diagnostického pohledu se v současnosti nejčastěji využívají molekuly tři – tzv. S-100 protein, CD1a a langerin. Protože se v literatuře o LCH běžně nesetkáváme se specifikací těchto molekul a v praktickém využití se situace většinou natolik zjednodušuje, že se přebírají jen názvy a výsledky stanovení protilátek v imunohistochemickém vyšetření, považujeme za vhodné seznámit zájemce s každou z těchto molekul blíže.
Do diagnostického procesu se jednotlivé molekuly zapojovaly postupně. Z tohoto pohledu je nejdéle známá skupina S-100 proteinů (80. léta 20. století), za ní následovala charakterizace molekuly CD1a (převážně 90. léta 20. století). Objevení langerinu a jeho využití v diagnostice je poměrně nové. Průkaz langerinu se začal uplatňovat až po roce 2000. S dostupností komerčních protilátek použitelných na formolem fixovaných tkání je postupně vytlačována ultrastrukturální diagnostika, která používá méně dostupnou a časově náročnější elektronovou mikroskopii.
S-100 proteiny
Jak již bylo zmíněno výše, k diagnostickému průkazu Langerhansových buněk se používá stanovení exprese takzvaného S-100 proteinu. Pod označením S-100 se však skrývá řada různých molekul. Proteiny S-100 jsou malé kyselé proteiny o molekulové hmotnosti 10 – 12 kD. První člen této rodiny byl identifikován již v roce 1965. Jednalo se o subcelulární frakci izolovanou z hovězího mozku. Jelikož součásti této frakce byly rozpustné ve 100% nasyceném roztoku síranu amonného při neutrálním pH, byla tato frakce pojmenována S-100 – „soluble in 100 % saturated ammonium sulphate at neutral pH“. Tento popisný název se natolik vžil, že je běžně používán do současnosti [36]. Mezi S-100 proteiny patří molekuly kódované v klastru v oblasti 1. chromozomu 1q21 – ty jsou označovány písmenem A a dále arabskou číslovkou (S-100 A1 – A16). Dalšími členy „S-100 rodiny“ jsou proteiny kódované geny ležící mimo oblast 1q21. Ty jsou pojmenovány pouze S-100 a následně písmenem bez číslice – S-100B (21q22.3), S-100G (Xp22.2), S-100P (4p16) a S-100Z (5q13.3) [37]. S-100 proteiny se podílejí na regulaci buněčných dějů, a to intracelulárních i extracelulárních (např. na progresi buněčným cyklem nebo na diferenciaci). Jsou lokalizovány v cytoplazmě a/ nebo v jádře širokého spektra různých typů buněk. Navíc jsou také prokazovány v extracelulárním prostoru, kde účinkují podobně jako cytokiny – přes receptory. Kromě vysoké afinity k vápenatým iontům vykazuje řada S-100 proteinů také afinitu k iontům zinečnatým a měďnatým, což může ovlivňovat jejich aktivitu v extracelulárním prostoru [38]. Langerhansovy buňky exprimují S-100B protein [39,40]. Kromě silné exprese S-100B proteinu je u Langerhansových buněk popisován také slabý stupeň exprese S-100A6 proteinu [41]. Na našem pracovišti používáme pro průkaz exprese skupiny S-100 proteinů u LCH v histologických řezech z formolem fixovaných a do parafinu zalitých tkání králičí polyklonální protilátku anti-S-100 (Dako) (obr. 7).
Molekula CD1a
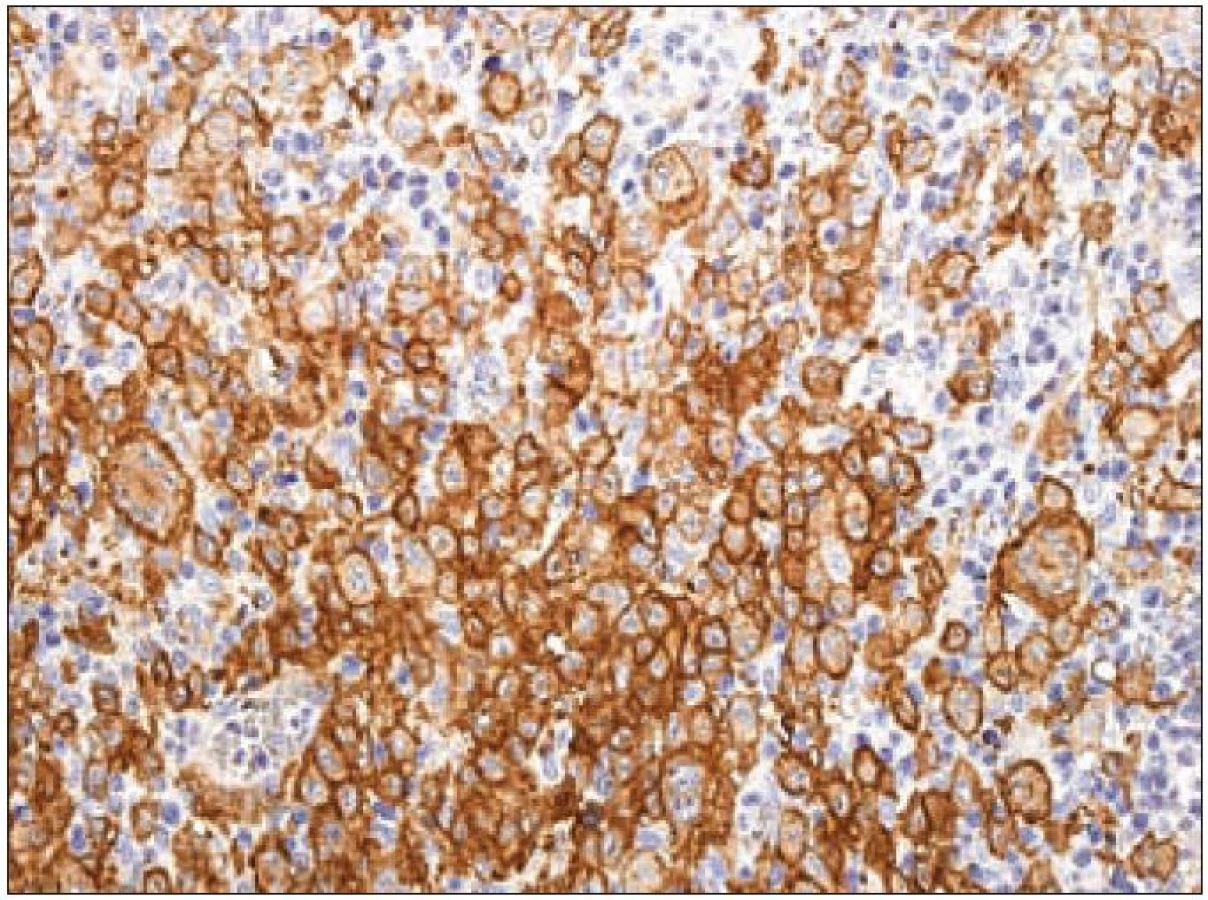
Další molekulou, jejíž průkaz se používá pro identifikaci Langerhansových buněk, je CD1a. CD1a má řadu synonymických názvů [37], ale označení podle CD klasifikace (cluster of differentiation) je nejpoužívanější. Molekula patří do rodiny transmembránových glykoproteinů, které jsou strukturálně podobné MHC proteinům I. třídy („major histocompatibility complex“). CD1 proteiny zprostředkovávají prezentaci primárně lipidových a glykolipidových antigenů vlastního nebo mikrobiálního původu pro T-lymfocyty a NK buňky. Lidský genom obsahuje 5 izoforem CD1. Dělí se na skupinu 1 (CD1a, b, c, e) a na skupinu 2 (CD1d). Geny jsou organizované v klastrech na 1. chromozomu. Členové rodin se pravděpodobně liší buněčnou lokalizací a specifitou pro konkrétní lipidové ligandy. Gen CD1A se nachází na dlouhém raménku prvního chromozomu 1q22 - q23. Protein CD1a (molekulová hmotnost 37 kDa) je lokalizován na buněčné membráně a na vezikulech časného endocytického systému. CD1a exprimují kortikální thymocyty, Langerhansovy buňky a další typy dendritických buněk [42]. Molekula CD1a, která nemá internalizační motiv s tyrozinem, je internalizována do časného endozomu mechanizmem nezávislým na systému klatrin/ dynamin (klatrinové vezikuly jsou v buňkách hlavními transportéry pro materiál pohlcený endocytózou). CD1a se recykluje zpátky na buněčný povrch mechanizmem závislým na malých GTPázách [43,44]. Na našem pracovišti používáme pro průkaz exprese molekuly CD1a u LCH v histologických řezech z formolem fixovaných a do parafinu zalitých tkání myší monoklonální protilátku proti lidské molekule CD1a (klon O10, Immunotech/ Beckman Coulter) (obr. 8).
Langerin
Jak jsme zmínili, dendritické buňky se dělí do několika skupin. Jedním z kritérií je dělení podle exprese C-typu lektinů. Lektiny C-typu jsou proteiny dependentní na vápenatých iontech, které vážou glykany. Jsou homologní v primární a sekundární struktuře oblasti pro vazbu cukru („carbohydrate-recognition domain“ – CRD). C-typy lektinů tvoří homodimery, heterodimery a oligomery. Liší se v typu glykanu, který jsou schopny s vysokou afinitou rozeznat. Tyto proteiny slouží jako receptory adheze a signální receptory při mnoha funkcích imunitního systému (imunita zánětů, nádorů a virových infekcí) [45]. Ze skupiny lektinů C-typu exprimují Langerhansovy buňky tzv. langerin [46]. Langerin (molekulová hmotnost 37 kD) je transmembránový protein II. typu s extracelulární CRD doménou. Rozpoznává a váže manózu, fukózu a N-acetylglukozamin. Langerin je synonymicky nazýván CD207 („Cluster of Differentiation 207“), nebo také „C-type lectin domain family 4 member K“, CLEC4K nebo „Langerhans cell specific C-type lectin“ [37]. Langerin je exprimován pouze v Langerhansových buňkách, ve kterých je lokalizován v Birbeckových granulech. Gen CD207 se nachází na druhém chromozomu, v oblasti 2p13. Vazba manózy na langerin vede k internalizaci antigenu nesoucího manózu procesem, který připomíná „zazipování“ do membránových struktur Langerhansovy buňky. Spojováním membrán vznikají Birbeckova granula [47]. Ke zpracování antigenu dochází tedy variantní cestou, odlišnou od klasické cesty zprostředkované makrofágy. Extracelulární doména langerinu, a zvláště CRD, má klíčovou roli v zipování membrány při tvorbě Birbeckových granul (důležitou roli hraje i cytoplazmatická doména). Langerin ovlivňuje nejen tvorbu Birbeckových granul, ale také celkové morfologické změny Langerhansových buněk [48]. Na základě studií s langerinem vyplynulo, že Birbeckova granula mohou vznikat jak z buněčné membrány, tak z membrán Golgiho systému. Identifikace langerinu jako receptoru pro endocytózu indukujícího tvorbu Birbeckových granul, ukazuje, že Birbeckova granula fungují v dráze zpracování antigenů [47]. Pro průkaz exprese langerinu v histologických řezech z formolem fixovaných a do parafinu zalitých tkání na našem pracovišti používáme v současnosti myší monoklonální protilátku proti lidskému langerinu (klon 12D6, Novocastra/ Leica) (obr. 9).

Molekula CD68
Expresí S-100B proteinu, langerinu a molekuly CD1a se Langerhansovy buňky odlišují od tkáňových makrofágů, u kterých se exprese ani jednoho z těchto proteinů neprokáže. Makrofágy jsou naproti tomu charakteristické silnou pozitivitou molekuly CD68, která je v Langerhansových buňkách prokazována variabilně nebo je negativní (obr. 10).

Ki-67
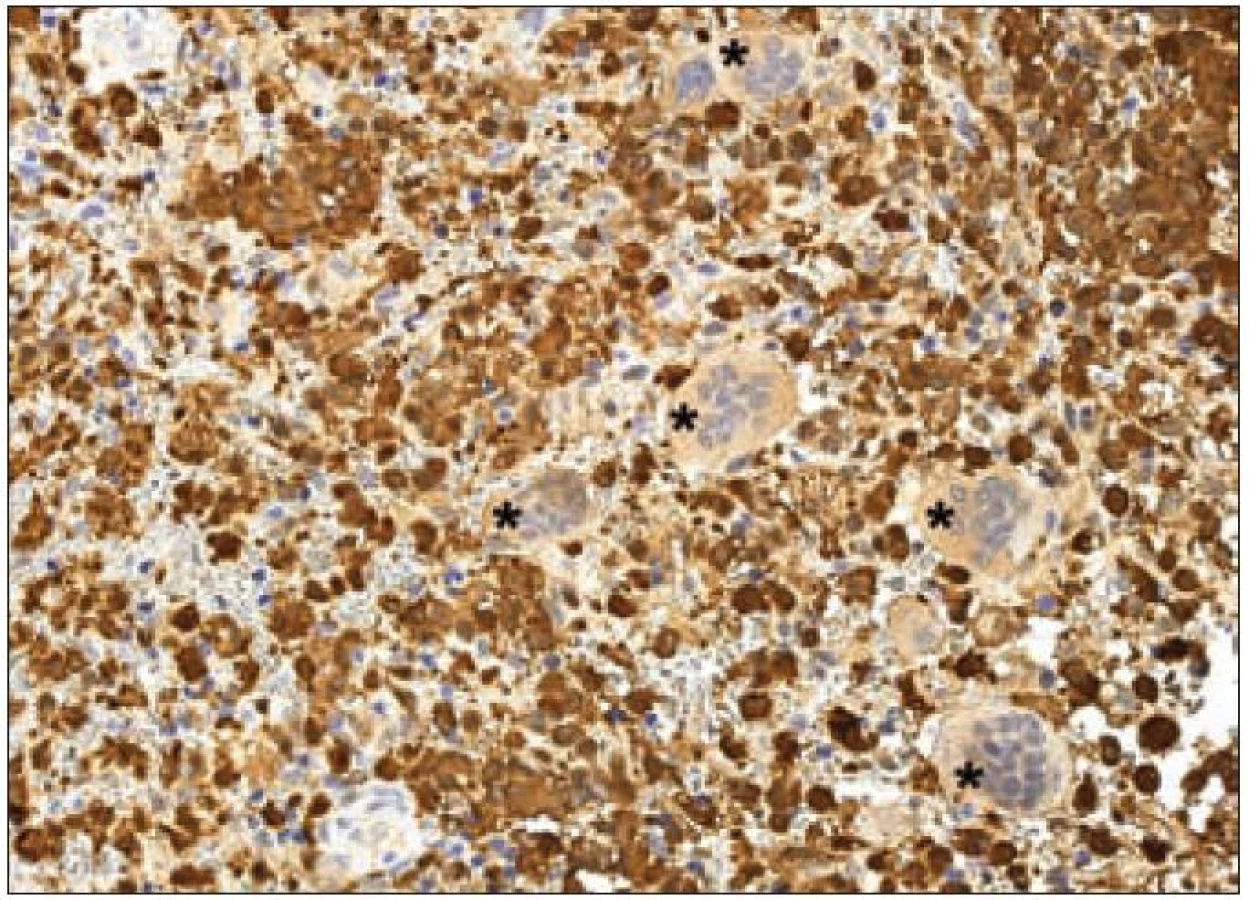
Z praktického hlediska má význam stanovení proliferační aktivity Langerhansových buněk v rámci různých forem LCH. Kromě sledování mitotické aktivity je v posledních letech dostupné vyšetření řady molekul buněčného cyklu, které lze k posouzení proliferační frakce různých patologických procesů využít. Z rutinně využívaných markerů se ujala detekce proliferačního markeru Ki-67 [23]. Molekula Ki-67 (synonymicky „antigen identified by monoclonal antibody Ki-67“, molekulová hmotnost 359 kD) je kódována genem MKI67, který se nachází na 10. chromozomu v oblasti 10q25 - qter. Název tohoto proteinu vznikl spojením počátečních písmen města, kde byla protilátka produkována a popsána (Univerzita v Kielu v Německu) a čísla „67“ originálního klonu v 96jamkové destičce [49]. Ki-67 je jaderný nehistonový protein, který je nezbytný pro proliferaci buněk. Přestože je popsána sekvence molekuly a mnoho jejích vlastností, je zatím velmi málo známo o její funkci v buňce. Ki-67 je exprimována v G1, S, G2 a M fázi buněčného cyklu. Během interfáze je molekula Ki-67 detekována relativně homogenně v jádře, zatímco v mitóze je většina proteinu lokalizována na povrchu chromozomů. Hladina exprese proteinu Ki-67 kolísá v průběhu buněčného cyklu. Je nízká v G1 a S fázi, maximum exprese dosahuje v M fázi. Po vstupu do klidové fáze (G0) je molekula Ki-67 rychle degradována a není exprimována. Na rozdíl od mnoha jiných proteinů spojených s regulací buněčného cyklu a používaných pro odhad stupně mitotické aktivity se molekula Ki-67 neúčastní opravných procesů DNA. Z těchto důvodů se exprese molekuly Ki-67 velmi dobře hodí ke sledování proliferační aktivity buněk a je také k těmto účelům nejčastěji používána v diagnostické praxi i v experimentálních podmínkách. Protilátka Ki-67 nebyla vhodná pro použití pro formolem fixované tkáně. Teprve později byly vyvinuté nové protilátky pro identifikaci exprese Ki-67, které byly schopny se navázat na Ki-67 antigen i po formolové fixaci (MIB-1 a MIB-3). V současnosti je známo několik protilátek pro detekci exprese Ki-67, ale nejčastěji se používá monoklonální myší protilátka MIB-1. Výsledek imunohistochemické reakce s protilátkou Ki-67/ MIB-1 je hodnocen procentuálním zastoupením pozitivních nádorových buněk. Proliferační frakce posuzovaná podle pozitivity Ki-67 je u LCH variabilní a podle různých autorů kolísá od 3 % do 25 % Langerhansových buněk [50]. Paradoxně je často pozorováno pouze malé množství mitóz, jak jsme již uvedli. Významně vyšší procento Ki-67 pozitivních Langerhansových buněk bylo zaznamenáno u nemocných s multiorgánovým onemocněním než u forem monoorgánových [51]. Využitelnost posouzení proliferační aktivity u LCH je však stále málo prozkoumaná. Z našich zkušeností vyplývá, že vyšší proliferační obrat např. u monoorgánových forem může signalizovat schopnost progrese onemocnění, recidivy nebo potenciální progresi a diseminaci (obr. 11). Konzultačně jsme vyšetřovali lymfatické uzliny u 26letého muže s krční lymfadenopatií, u kterého četné Langerhansovy buňky vykazovaly mitotickou aktivitu a Ki-67/ MIB-1 bylo pozitivní v 10 % a fokálně až ve 30 % nádorové populace. Z toho důvodu jsme se obávali progrese nemoci. Ta měla skutečně progresivní průběh s postižením dalších lymfatických uzlin. Langerin byl v tomto případě pozitivní prakticky v celé populaci patologických Langerhansových buněk, nález signalizující nezralost těchto buněk a pravděpodobný blok diferenciace. Využití proliferačních markerů u LCH bude nepochybně oceněno teprve s průběhem analýzy většího množství případů.

Sarkom z Langerhansových buněk
Kromě typické histiocytózy z Langerhansových buněk WHO klasifikace oficiálně vyčleňuje případy s atypickými Langerhansovými buňkami odpovídající nádoru vysokého stupně malignity – sarkom z Langerhansových buněk [2]. Jde o vzácné onemocnění, se kterým sami nemáme zkušenosti. Do roku 2007 bylo zaznamenáno 20 případů s přijatelně charakterizovanou nádorovou populací [52]. Sarkom z Langerhansových buněk postihuje dospělé osoby, častěji ženy. Vyskytuje se v kůži a v měkkých tkáních, při multiorgánové manifestaci postihuje lymfatické uzliny, játra, slezinu, kosti a plíce. Při diagnóze bývá onemocnění často již v pokročilém stadiu. Nádorové buňky mají charakteristiky pleomorfního maligního nádoru s vysokým mitotickým obratem. K identifikaci buněk je nutné použít imunohistochemické vyšetření, jehož výsledky jsou svým spektrem průkazu antigenů obdobné jako u klasické LCH, avšak exprese markerů jsou uváděné jako fokální a slabší. Ultrastrukturální vyšetření má význam v průkazu Birbeckových granul, pokud se neaplikuje protilátka proti langerinu. Z tohoto pohledu, i když máme jen zprostředkované znalosti o obrazu této formy LCH, lze usuzovat, že případy LCH s vysokou proliferační frakcí u dospělých by mohly být blízké sarkomu z Langerhansových buněk. Sarkom by pak mohl být vystupňovanou formou s vysokou proliferační aktivitou za současné ztráty buněčné kontroly reparativních mechanizmů a apoptotické aktivity Langerhansových buněk [53].
Diferenciální diagnóza histiocytózy z Langerhansových buněk
Vzhledem k širokému spektru věkového výskytu LCH je i histopatologická diferenciální diagnóza rozmanitá. V raném dětském věku je třeba při postižení kůže odlišit seborrhoickou dermatitidu, juvenilní xantogranulom a případně tak zvané self-healing histiocytózy [54], u dospělých např. kožní formy lymfomů – lymfomatoidní papulóza/ anaplastický velkobuněčný lymfom (ALCL) nebo mycosis fungoides. Při uzlinovém postižení je nutné vyloučit reaktivní hyperplazie Langerhansových buněk (např. při drenáži nádorových onemocnění do spádových lymfatických uzlin [55]), reaktivní sinusovou histiocytózu u zánětů, tzv. sinusovou histiocytózu s masivní lymfadenopatií (syndrom Rosai-Dorfman) [56], granulomatózní lymfadenitídy a z maligních onemocnění zejména klasickou formu Hodgkinova lymfomu, ALCL a sarkomy z dendritických buněk. U kostních projevů je v chronickém stadiu LCH s výskytem pěnitých makrofágů možná záměna s chronickou hnisavou osteomyelitidou. Z našich zkušeností s LCH v kostní oblasti víme, že je možná záměna s chondroblastomem (S-100 protein pozitivní), zejména při biopsiích malého rozsahu bez přítomnosti diferencující se chrupavky. Samostatnou kapitolu představují plicní histiocytózy z Langerhansových buněk u dospělých pacientů, kuřáků, které mají polyklonální povahu a reaktivní charakter [57,58]. Pře-hled diferenciální diagnózy LCH podává tab. 2.

Závěr
Přes pokroky v charakterizaci Langerhansových buněk a v diagnostice LCH zůstává stále otevřené pole k řešení otázek, jež se rodí s novými poznatky. Není dosud jasná především sama podstata nemoci. Nevíme, zda jde o klonální onemocnění primárně vznikající na základě mutačních kroků nebo zda je LCH chorobou vznikající deregulací Langerhansových buněk antigenními stimuly, následným růstem populace Langerhansových buněk a s klonálním zvratem až v další fázi vývoje choroby. Dosud známé změny na chromozomech mohou být pouze druhotné. Etiologie je tedy také stále nejasná. Zůstává otevřená otázka proliferace Langerhansových buněk a faktory jejich lokální stabilizace v monoorgánové podobě (kostní, uzlinové, kožní) a faktory, které umožňují diseminaci do lymforetikulárních a hemopoeticky aktivních tkáni ve formách multiorgánových. Není také jasný tzv. homing fenomén buněk Langerhansovy histiocytózy, a tím zůstává otevřená otázka, jaké faktory umožní např. kolonizaci plic v rámci systémové LCH dětí a naopak, proč tyto buňky v rámci generalizace neprokazujeme např. v ledvinách. Je pravděpodobné, že se také bude lépe definovat vztah klasické LCH a sarkomu z Langerhansových buněk a přechodné formy – viz zmíněný případ pacienta s LCH s vysokou proliferační frakcí. Z hlediska hledání nových přístupů k léčbě nabývá na významu přesná diagnostika a charakterizace buněk LCH (obdobně jako u jiných onkologických onemocnění). Patolog v této situaci musí hledat cesty nejen ve zjemnění diagnózy s charakterizací Langerhansových buněk, ale ve spolupráci s imunology je třeba zkoumat také vztahy mezi subpopulacemi těchto buněk (např. langerin pozitivní vs negativní) a vztahy cytokinové komunikace mezi buňkami dendritickými, tkáňovými histiocyty a osteoklasty a mezi populacemi lymfoidních buněk. Zde by mohl být klíč k rozpoznání chování této „záhadné“ histiocytózy, původně koncipované jako „neznámá veličina“ – histiocytóza X. I když mnohé otázky zůstávají zatím nezodpovězeny, je možné říci, že rekapitulace poznatků více než jednoho století zájmu o tuto chorobu ukazuje, že se naše poznání posunulo podstatně kupředu.
Práce byla podpořena projekty MZO FNM 2005/ 6704 a MSM 0021620813.
prof. MUDr. Roman Kodet, CSc.
www.lf2.cuni.cz
e-mail: roman.kodet@lfmotol.cuni.cz
Doručeno do redakce: 9. 9. 2010
Zdroje
1. Adam Z, Pour L, Krejčí M et al. Histiocytóza z Langerhansových buněk u osob dospělého věku – nemoc s mnoha tvářemi. Zkušenosti jednoho pracoviště a přehled projevů nemoci. Vnitř Lék 2008; 54 : 1063 – 1080.
2. Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 2nd ed. Lyon: IARC Press 2008.
3. Adam Z, Krejčí M, Pour L. Histiocytární choroby. Vnitř Lék 2009; 55 : 109 – 124.
4. Fraser J. Skeletal lipoid granulomatosis (Hand-Shüller-Cristian’s Disease). Br J Surg 1935; 22 : 800 – 824.
5. Abt AF, Denenholz EJ. Letterer-Siwe’s disease: Splenohepatomegaly associated with widespread hyperplasia of nonlipoid-stroing macrophages. Discussion of the so-called reticulo-endothelioses. Am J Dis Child 1936; 51 : 499 – 522.
6. Otani S, Ehrlich JC. Solitary granuloma of bone: simulating primary neoplasm. Am J Pathol 1940; 16 : 479 – 490.
7. Lichtenstein L, Jeffe HL. Eosinophilic granuloma of bone: with report of a case. Am J Pathol 1940; 16 : 595 – 604.
8. Wallgren A. Systemic reticuloendothelial granuloma: Nonlipod reticuloendotheliosis and Schuller-Christian disease. Am J Dis Child 1940; 60 : 471 – 500.
9. Farber S. The nature of “solitary eosinophilic granuloma” of bone. Am J Pathol 1941; 17 : 625 – 629.
10. Green WT, Farber S. “Eosinophilic or solitary granuloma” of bone. J Bone Joint Surg 1942; 24 : 499 – 526.
11. Mallory TB. Pathology: Diseases of Bone. N Engl J Med 1942; 227 : 955 – 960.
12. Laymon CW, Sevenants JJ. Systemic reticuloendothelial granuloma; comparison of Letterer-Siwe disease, Schueller-Christian disease and eosinophilic granuloma. Arch Derm Syphilol 1948; 57 : 873 – 890.
13. Dennis JW, Rosahn PD. The primary reticulo-endothelial granulomas, with report of an atypical case of Letterer-Siwe’s disease. Am J Pathol 1951; 27 : 627 – 653.
14. Lichtenstein L. Histiocytosis X, Integration of eosinophilic: granutoma of bone, “Letterer-Siwe disease”, and “Schüller-Christian disease” as related manifestations of a single nosologic entity. Arch Pathol 1953; 56 : 84 – 102.
15. Birbeck MS, Breathnach AS, Everall JD. An electron microscope study of nasal melanocytes and high‑level clear cells (Langerhans cells) in vitiligo. J Invest Dermat 1961; 37 : 51 – 63.
16. Basset F, Turiaf MJ. Identification par la microscopie electronique de particules de nature probablement virale dans les liaisons granulomateuses d’une histiocytose “X” pulmonaire. C R Acad Sci Hebd Seances Acad Sci D 1965; 261 : 3701 – 3703.
17. Basset F, Nezelof C, Turiaf J. Presence en microscopie electronique de structures filamenteuses originales dans les lesions pulmonaires et osseuses de l’histiocytose x. Etat actuel de la question. Bull Mem Soc Med Hop Paris 1966; 117 : 413 – 426.
18. Nezelof C, Basset F, Rousseau MF. Histiocytosis X histogenetic arguments for a Langerhans cell origin. Biomedicine 1973; 18 : 365 – 371.
19. Nezelof C, Barbey S. Histiocytosis: nosology and pathobiology. Pediatr Pathol 1985; 3 : 1 – 41.
20. Yu RC, Chu C, Buluwela L et al. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994; 343 : 767 – 768.
21. Willman CL, Busque L, Griffith BB et al. Langerhans’-cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med 1994; 331 : 154 – 160.
22. Yousem SA, Colby TV, Chen YY et al. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol 2001; 25 : 630 – 636.
23. Schouten B, Egeler RM, Leenen PJ et al. Expression of cell cycle‑related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2002; 24 : 727 – 732.
24. Betts DR, Leibundgut KE, Feldges A et al. Cytogenetic abnormalities in Langerhans cell histiocytosis. Br J Cancer 1998; 77 : 552 – 555.
25. Murakami I, Gogusev J, Fournet JC et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol 2002; 33 : 555 – 560.
26. Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet 1987; 1 : 208 – 209.
27. da Costa CE, Annels NE, Faaij CM et al. Presence of osteoclast‑like multinucleated giant cells in the bone and nonostotic lesions of Langerhans cell histiocytosis. J Exp Med 2005; 201 : 687 – 693.
28. Kodet R, Elleder M, Šmelhaus V et al. Diseminovaná histiocytosis X. Česk Patol 1984; 20 : 19 – 26.
29. Williams JW, Dorfman RF. Lymphadenopathy as the initial manifestation of histiocytosis X. Am J Surg Pathol 1979; 3 : 405 – 421.
30. Favara B. Histopathology of the liver in histiocytosis syndromes. Pediatr Pathol Lab Med 1996; 16 : 413 – 433.
31. Kodet R, Zítková M. Morfologické a rentgenologické plicní nálezy pri diseminované histiocytóze X. Česk Pediatr 1985; 40 : 634 – 638.
32. Mierau GW, Favara BE, Brenman JM. Electron microscopy in histiocytosis X. Ultrastruct Pathol 1982; 3 : 137 – 142.
33. Elleder M. Activity of alpha-d-mannosidase in human Langerhans epidermal cells. Virchows Arch B Cell Pathol 1975; 19 : 93 – 96.
34. Novak N, Gros E, Bieber T et al. Human skin and oral mucosal dendritic cells as ‘good guys’ and ‘bad guys’ in allergic immune responses. Clin Exp Immunol 2010; 161 : 28 – 33.
35. Toebak MJ, Gibbs S, Bruynzeel DP et al. Dendritic cells: biology of the skin. Contact Dermatitis 2009; 60 : 2 – 20.
36. Salama I, Malone PS, Mihaimeed F et al. A review of the S-100 proteins in cancer. Eur J Surg Oncol 2008; 34 : 357 – 364.
37. Safran M, Dalah I, Alexander J et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010: Print 2010.
38. Marenholz I, Heizmann CW, Fritz G. S-100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 2004; 322 : 1111 – 1122.
39. Park HR, Min SK. Expression of S-100A2 and S-100B proteins in epithelial tumors of the skin. J Cutan Pathol 2003; 30 : 373 – 378.
40. Nishikawa Y, Sato H, Oka T et al. Immunohistochemical discrimination of plasmacytoid dendritic cells from myeloid dendritic cells in human pathological tissues. J Clin Exp Hematop 2009; 49 : 23 – 31.
41. Shrestha P, Muramatsu Y, Kudeken W et al. Localization of Ca(2+)‑binding S-100 proteins in epithelial tumours of the skin. Virchows Arch 1998; 432 : 53 – 59.
42. Krenács L, Tiszalvicz L, Krenács T et al. Immunohistochemical detection of CD1a antigen in formalin‑fixed and paraffin‑embedded tissue sections with monoclonal antibody. J Pathol 1993; 171 : 99 – 104.
43. Salamero J, Bausinger H, Mommaas AM et al. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. J Invest Dermatol 2001; 116 : 401 – 408.
44. Salio M, Silk JD, Cerundolo V. Recent advances in processing and presentation of CD1 bound lipid antigens. Curr Opin Immunol 2010; 22 : 81 – 88.
45. Varki A, Cummings RD, Esko JD et al. eds. Essentials of Glycobiology, Cold Spring Harbor. 2nd ed. New York: Cold Spring Harbor Laboratory Press 2009.
46. Valladeau J, Duvert-Frances V, Pin JJ et al. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur J Immunol 1999; 29 : 2695 – 2704.
47. Valladeau J, Ravel O, Dezutter-Dambuyant C et al. Langerin, a novel C‑type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000; 12 : 71 – 81.
48. Thépaut M, Valladeau J, Nurisso A et al. Structural studies of langerin and Birbeck granule: a macromolecular organization model. Biochemistry 2009; 48 : 2684 – 2698.
49. Gerdes J, Schwab U, Lemke H et al. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer 1983; 31 : 13 – 20.
50. Egeler RM, van Halteren AG, Hogendoorn PC et al. Langerhans cell histiocytosis: fascinating dynamics of the dendritic cell-macrophage lineage. Immunol Rev 2010; 234 : 213 – 232.
51. Amir G, Weintraub M. Association of cell cycle‑related gene products and NF-kappaB with clinical parameters in Langerhans cell histiocytosis. Pediatr Blood Cancer 2008; 50 : 304 – 307.
52. Bohn OL, Ruiz-Argüelles G, Navarro L et al. Cutaneous Langerhans cell sarcoma: a case report and review of the literature. Int J Hematol 2007; 85 : 116 – 120.
53. Bank MI, Rengtved P, Carstensen H et al. p53 expression in biopsies from children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2002; 24 : 733 – 736.
54. Kodet R, Elleder M, De Wolf-Peeters C et al. Congenital histiocytosis: a heterogeneous group of diseases, one presenting as so-called congenital self-healing histiocytosis. Pathol Res Pract 1991; 187 : 458 – 466.
55. Safali M, McCutcheon JM, Wright DH. Langerhans cell histiocytosis of lymph nodes: draining a papillary carcinoma of the thyroid. Histopathol 1997; 30 : 599 – 603.
56. Kodetová D, Kodet R, Syrůček M et al. Sinusová histiocytóza s masivní lymfadenopatií – diseminovaná forma syndromu Rosai-Dorfman. Česk Patol 1996; 32 : 53 – 59.
57. Abbott GF, Rosado-de-Christenson ML, Franks TJ et al. From the archives of the AFIP: pulmonary Langerhans cell histiocytosis. Radiographics 2004; 24 : 821 – 841.
58. Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27 : 1272 – 1285.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2010 Číslo Supplementum 2
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
Najčítanejšie v tomto čísle
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících