
Plazmocelulární leukemie
Plasma cell leukaemia
Plasma cell leukaemia (PCL) is a rare and aggressive variant of plasma cell dyscrasias characterized by the presence of circulating plasma cells. It is classified as either primary PCL occurring “de novo” or as secondary PCL in patients with relapsed/refractory myeloma. Primary PCL is a distinct clinicopathological entity with different cytogenetic and molecular findings. The clinical course is aggressive with short remissions and survival. The diagnosis is based upon the percentage (> 20%) and absolute number (2 x 109/l) of plasma cells in the peripheral blood. Induction therapy needs to begin promptly and to have high clinical activity leading to rapid disease control. Intensive chemotherapy regimens and bortezomib-based regimens are recommended followed by high-dose therapy with autologous stem cell transplantation if feasible. Allogeneic transplantation can be considered in younger patients. This work reviews recent knowledge of this blood malignancy with very poor prognosis.
Keywords:
plasma cell leukaemia, transplantation, myeloma, bortezomib
Autoři:
T. Jelínek; H. Plonková; R. Hájek
Působiště autorů:
Lékařská fakulta, Ostravská univerzita v Ostravě
; Klinika hematoonkologie, Fakultní nemocnice Ostrava
Vyšlo v časopise:
Transfuze Hematol. dnes,19, 2013, No. 3, p. 152-162.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Plazmocelulární leukemie (PCL) je vzácná a agresivní forma plazmocelulární dyskrazie charakterizována přítomností cirkulujících plazmocytů. Dělíme ji na primární PCL objevující se „de novo“ a na sekundární, která se vyskytuje u nemocných s relabovaným či refrakterním mnohočetným myelomem. Primární plazmocelulární leukemie je odlišná klinicko--patologická jednotka s rozdílnými cytogenetickými a molekulárními nálezy, než jsou u sekundární plazmocelulární leukemie. Klinický průběh je agresivní s krátkými remisemi a krátkou dobou přežití. Diagnóza je založena na nálezu více než 20 % plazmocytů v periferní krvi a jejich absolutním počtem více než 2 x 109/l. V současné době se diskutuje o přehodnocení diagnostických kritérií a hodnocení léčebné odpovědi. U primární plazmocelulární leukemie je léčbu nutné zahájit neprodleně, s použitím intenzivních chemoterapeutických režimů a režimů založených na bortezomibu, následované autologní transplantací kmenových buněk. U mladších nemocných je na zvážení i alogenní transplantace. Práce shrnuje současný stav znalostí a léčebných možností u tohoto zatím prognosticky významně nepříznivého krevního nádorového onemocnění.
Klíčová slova:
plazmocelulární leukemie, transplantace, myelom, bortezomib
ÚVOD
Plazmocelulární leukemie (PCL) je vzácná forma plazmocelulární dyskrazie, nejagresivnější z lidských monoklonálních gamapatií. Incidence tohoto onemocnění je velmi nízká a odhaduje se v evropské populaci na 0,04 případy na 100 000 obyvatel za rok (1), respektive se objeví asi u 2–4 % nemocných s mnohočetným myelomem (MM) (2, 3). Stejně jako mnohočetný myelom je i PCL častější u Afroameričanů než u kavkazské populace (4). Definice PCL, která je stále platná, byla vytvořena v sedmdesátých letech minulého století. Mezi její tvůrce patří R. Kyle, jeden z nestorů americké hematologie. Na základě jeho informací (osobní komunikace) lze konstatovat, že šlo o zcela intuitivní empirické stanovení arbitrážní hodnoty procent plazmocytů v periferní krvi. Tato kritéria jsou platná dodnes, třebaže je dnes výrazná snaha upravit kritéria na základě nejnovějších poznatků (5). Plazmocelularní leukemie je definována přítomností více než 20 % plazmocytů v periferní krvi, přičemž musí být absolutní počet plazmocytů nad 2 x 109/l (6, 7). Existuje de novo primární PCL a sekundární PCL vyskytující se u nemocných s pokročilým a refrakterním MM. Primární a sekundární PCL jsou dvě rozdílné, třebaže klinicky a biologicky podobné jednotky, které sdílejí obraz plazmocytů v periferní krvi a neutěšený klinický výstup. Primární PCL má vysokou citlivost (až 100%) na podanou agresivní chemoterapii. Nepříznivá prognóza je daná agresivitou onemocnění, a tudíž časnou reaktivací nádorové aktivity (8). Naopak sekundární PCL, kdy dochází k leukemizaci MM, představuje refrakterní terminální stav reagující na léčbu v méně než polovině případů, zpravidla rychle směřující k terminálnímu stavu nemoci. Cílem tohoto přehledného sdělení je poskytnout ucelený pohled na definici PCL, kritéria léčebné odpovědi a léčebná doporučení pro primární i sekundární PCL.
PŘÍTOMNÉ KLINICKÉ PROJEVY
Vzhledem k relativně nízké incidenci a prevalenci tohoto onemocnění pochází většina klinických dat z jednotlivých kazuistik a malých retrospektivních studií (9, 3, 10). Zatím nebyly publikovány žádné prospektivní studie. Hlavní klinické a laboratorní znaky, léčebné odpovědi a přežití nemocných s primární PCL jsou shrnuty v tabulce 1.
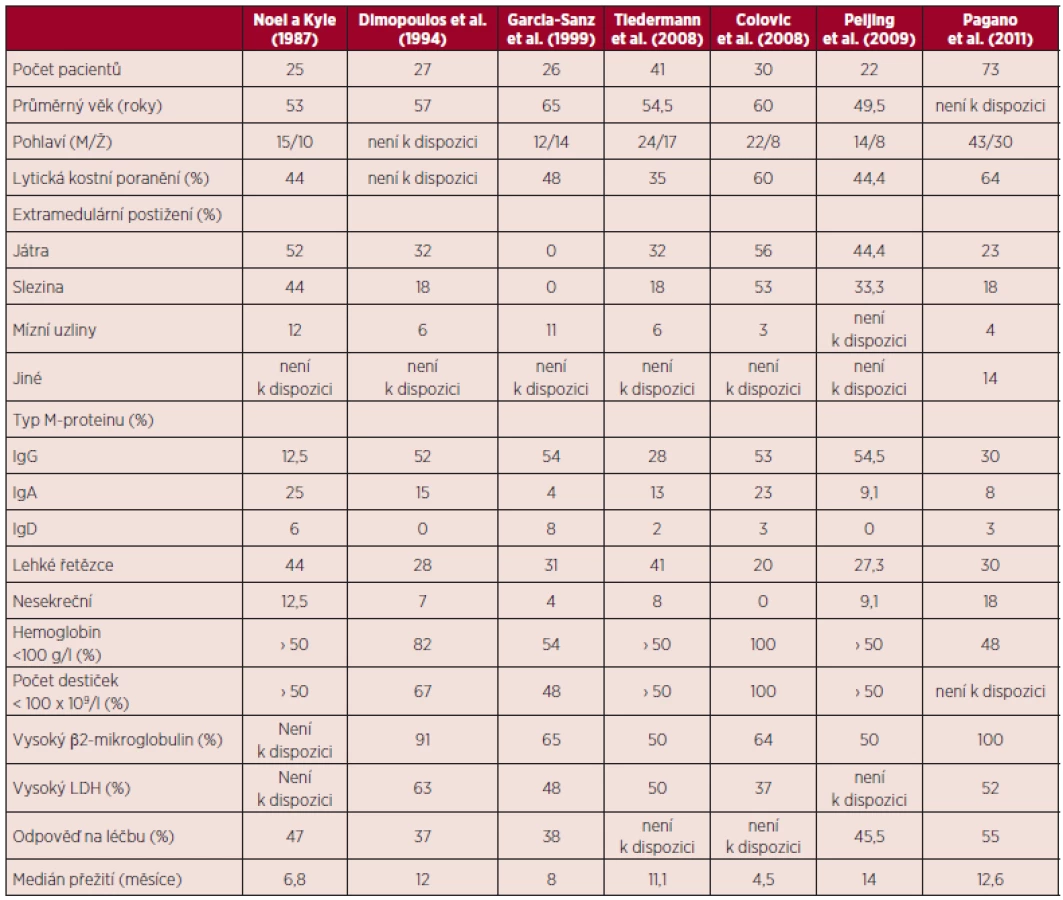
Průměrný věk nemocných s primární PCL je asi o 10 let nižší než průměrný věk (65 let) běžné myelomové populace (11) a nemocných se sekundární PCL (3). Stejně jako u akutních leukemií je čas od počátku vzniku onemocnění do rozvoje prvních příznaků velmi krátký (týdny). U nemocných s PCL jsou oproti nemocným s MM obvykle častější a závažnější příznaky související s těžkou anémií (bledost, dušnost) nebo hemoragickou diatézou vyplývající z trombocytopenie. Důvodem je zpravidla extenzivní infiltrace kostní dřeně plazmocyty s anaplastickou a plazmablastovou morfologií (obr. 1A).

V souladu s agresivním klinickým průběhem má vyšší podíl nemocných s primární PCL významnou leukocytózu a zvýšenou hladinu laktátdehydrogenázy a ß2-mikroglobulinu oproti nemocným s MM (12). Přítomnost osteolytických ložisek je nižší než u MM (u primární PCL 35 %, u sekundární PCL 53 %) (3), což pravděpodobně souvisí s agresivitou a rychlostí vývoje onemocnění. Při klinickém vyšetření můžeme u nemocných ve zvýšené míře pozorovat i hepatosplenomegalii, lymfadenopatii, nález na plících spojený s pleurálním výpotkem, neurologický deficit při postižení centrálního nervového systému a extramedulární plazmocytové měkkotkáňové infiltráty. Velký podíl nemocných s PCL secernuje jen lehké řetězce na rozdíl od klasického myelomu (26–44 % vs. 15 %) (11). Lékaři si musí být vědomi možného syndromu nádorového rozpadu při velké nádorové mase a zvýšeném proliferativním indexu.
DIAGNOSTICKÁ KRITÉRIA
Původní diagnostická kritéria PCL stanovena arbitrárně zahrnují více než 20 % cirkulujících plazmocytů a absolutní počet plazmocytů v periferní krvi nad 2 x 109/l (6). Tato kritéria zatím nebyla nikdy prospektivně ověřena. Není jisté, zda přesně odlišují PCL od MM. Kritéria jsou platná právě při použití morfologického hodnocení. Zvláště s ohledem na možnosti stanovení nádorových plazmocytů průtokovou cytometrií v periferní krvi je pravděpodobné, že i nižší počet plazmoblastů již může znamenat vývoj diagnózy PCL.
Dodnes není zcela porozuměno kontrolním mechanismům, díky kterým zůstávají plazmocyty převážně v kostní dřeni a do krevního řečiště se dostávají pouze výjimečně. Malý poměr plazmocytů je detekován u nemocných s celou řadou plazmocelulárních dyskrazií včetně nově diagnostikovaného MM, doutnajícího MM a výjimečně i u monoklonální gamapatie nejasného významu (MGUS) (13, 14). Od nádorových stavů je vždy nutné diferenciálně diagnosticky odlišit přítomnost zvýšeného počtu polyklonálních plazmocytů v periferní krvi u nenádorových onemocnění, jako je vážná sepse, infekční mononukleóza či sérová nemoc (15, 16).
Není rovněž jednoznačné, zda potřebujeme obě dvě kritéria. V mnoha studiích se považovalo za dostatečné splnit jednu z podmínek k uzavření diagnózy PCL (> 20 % cirkulujících plazmocytů a absolutní počet vyšší než 2 x 109/l v periferní krvi). Například u nemocných vystavených intenzivnější léčbě s malou rezervou kostní dřeně a leukopenií bývá splněno jen procentuální kritérium, nikoliv však absolutní. Pravděpodobně stačí splnění pouze jednoho kritéria ke stanovení diagnózy této klinické jednotky. Správná a včasná diagnóza PCL je závislá na schopnosti hematologa rozpoznat plazmocyty v nátěru z periferní krve. Hematologové by si měli být vědomi klinické významnosti pečlivého morfologického vyšetření nátěrů periferní krve (obr. 1B), aby mohla být vyloučena přítomnost cirkulujících plazmocytů.

Řada myelomových expertů se domnívá, že by diagnostická kritéria měla být pozměněna. Názory na to jakým způsobem se však liší. Současná kritéria PCL, i když stačí splnit pouze jedno z nich, mohou podhodnotit skutečnou četnost tohoto onemocnění. Již přítomnost blastického typu plazmocytů v kostní dřeni je považována za prognosticky nepříznivý faktor (17). Přítomnost i malého množství cirkulujících plazmocytů je známkou vysoce proliferativního agresivního procesu. Je pravděpodobné, že může jít v řadě případů jen o časně zachycenou PCL, která bez podání adekvátní léčby může velmi rychle rozvinout obraz PCL. Snahou současných prospektivních studií je zjistit, jestli nižší hodnoty (např. 5 % či více plazmocytů v periferní krvi a/nebo absolutní počet plazmocytů v periferní krvi 0,5 x 109/l) má stejný prognostický efekt jako platná kritéria. Doposud nemáme k dispozici přídatná diagnostická kritéria odhalující časný PCL proces. Pečlivé vyšetření periferní krve konvenčním mikroskopem by mělo být provedeno u všech nemocných s MM, u kterých existuje klinické podezření na PCL, jako je leukocytóza či elevace laktátdehydrogenázy. Novější metody k detekci časné PCL, jako je průtoková cytometrie, by měly mít jednoznačně přednost. Jejich význam však musí být prospektivně zhodnocen v multicentrických studiích, a to nejlépe u nově diagnostikovaných nemocných. V tuto chvíli tedy neexistuje jednoznačný konsenzus ohledně přehodnocení diagnostických kritérií.
Imunofenotyp PCL
V porovnání se studiemi provedenými u MM jsou informace o imunofenotypu při stanovení diagnózy a při sledování zbytkového nádorového onemocnění u PCL limitované. Základní markery plazmocytů (CD38 a CD138) jsou stejně exprimovány jak u MM, tak u PCL. Některé znaky však mají rozdílnou expresi: u PCL je vyšší exprese CD20 antigenu (9) a nižší exprese antigenů CD9, CD117, CD56 a HLA-DR. Antigen CD28 související se zvýšenou proliferací plazmocytů je častěji exprimován u sekundární PCL (18). Zvýšená exprese CD27 u PCL je dávána do souvislosti s aktivací anti-apoptotické dráhy (19). Nově se ukazuje, že nadměrná exprese CD27 může vést k aktivaci NFκB vedoucí ke zvýšení anti-apoptotické aktivitě (20). Znak CD23 bývá asociován s přítomností translokace t(11;14) (21). Nejvýraznější rozdíl v imunofenotypu mezi nově diagnostikovaným MM a primární PCL je tedy v tom, že nádorové buňky primární PCL jsou méně často pozitivní pro CD56, CD71, CD117 a HLA-DR, ale častěji exprimují CD20, CD45, CD19, CD27 a CD23 (22). V tuto chvíli nelze zcela exaktně definovat typický imunofenotyp pro nově diagnostikovanou primární PCL.
Cytogenetické a genomické abnormality
Cytogenetické studie ukazují, že plazmocyty u PCL vykazují množství chromozomových odchylek. U více než 80 % nemocných s primární PCL jsou přítomny buňky s hypodiploidním nebo diploidním karyotypem oproti asi 40 % nemocných s MM (9). Delece chromozomu 13 a jeho monozomie jsou nejčastějším nálezem u PCL (10). Delece úseku 17p13.1 (gen p53) je významně četnější než u MM (u nové diagnózy 5–7 %), vyšší frekvence je u primární PCL oproti sekundární PCL (50 % vs. 75 %). Četnost IgH (14q32) translokací podle FISH analýz je podobná u obou typů PCL (87 % a 82 %). Četnost t(11;14) je u primárních PCL oproti MM vysoká (71 % vs. 15 až 20 %) (3). U MM patří t(11;14) mezi příznivé prognostické faktory. Nicméně vysoká prevalence této translokace u PCL naznačuje, že PCL je zásadně biologicky jiným onemocněním než MM. Na druhou stranu nebyly zaznamenány téměř žádné případy translokací t(4;14) nebo t(14;16). U PCL je rovněž častá amplifikace oblasti 1q21 (23). Pouze u 15 % primárních PCL je přítomna cMYC translokace, zatímco mutace N-Ras a K-Ras jsou zhruba stejně časté u PCL jako u MM (24). Epigenetické modifikace, jako např. p16 inaktivace, byly rovněž popsány u primární PCL (25, 26) stejně jako globální DNA hypometylace repetitivních genomových sekvencí (27). Při hodnocení genového expresního profilu (GEP) mělo překvapivě jen 44 % (16/27) nemocných s primární PCL vysoce rizikový profil obvyklý u myelomu s vysokým rizikem (GEP-70) (28). Fonseca et al. popisuje celogenomové sekvenování u jednoho nemocného se studiovými vzorky odebranými po celou dobu jeho nemoci včetně progrese MM do sPCL, kde podle zcela předběžných výsledků je dominantně patrný nárůst v počtu mutací v souvislosti s progredující nemocí (29). Lze stručně shrnout, že vyšetření chromozomových abnormalit lze považovat za vhodné u všech nově diagnostikovaných nemocných se suspektní PCL. Velká pozornost by měla být věnována nejčastějším změnám t(11;14), stejně jako abnormalitám chromozomů 1 a 17, zejména 1q+ a delece 17p. U sekundárních PCL můžeme často pozorovat komplexní změny karyotypu a dramatický vývoj mnohočetných chromozomových abnormalit ve srovnání s původním MM, jak demonstruje ukázka na obrázku 2. Další výzkum pomocí celogenomových metod je nutný k pochopení patogeneze primární PCL a transformace MM do sekundární PCL.

Na obrázku 2 jsou vzorky jsou odebrané od 52leté pacientky se stanovenou diagnózou MM, která měla v průběhu nemoci tři progrese, přičemž při třetí byl stav zhodnocen jako plazmocelulární leukemie (PCL). Došlo k infiltraci periferní krve abnormálními plazmocytárními buňkami a k vytvoření extramedulárního ložiska. Metodou array-CGH se podařilo získat celogenomové profily z období před poslední progresí (obr. 2A) a po ní (obr. 2B). Profil kostní dřeně po progresi do PCL a z extramedulárního ložiska se úplně shodují. Avšak genomové profily před progresí a po ní jsou odlišné – jde o změnu hyperdiploidního profilu na nonhyperdiploidní. Extramedulární ložisko vzniklo expanzí jednoho klonu nádorových plazmatických buněk z kostní dřeně, což potvrzuje shodný genomový profil obou testovaných vzorků (obr. 2B).

EXTRAMEDULÁRNÍ ŠÍŘENÍ
Současný nález extramedulárního šíření a PCL není neobvyklým nálezem. Mikroprostředí kostní dřeně hraje klíčovou roli v patogenezi MM tím, že spouští signální kaskády zprostředkující proliferaci myelomových buněk, jejich migraci a přežití, a tím vším se podílí na růstu myelomu a hnízdění maligních plazmocytů v kostní dřeni. Narušení těchto mechanismů může mít rozhodující vliv na unikátní biologické chování PCL (30). Dysbalance či nedostatek exprese adhezivních molekul na povrchu plazmocytů mohou být důležité. Častým nálezem je nepřítomnost antigenu CD56, neuronální adhezivní molekuly, která je podstatná pro ukotvení plazmocytů ve stromatu kostní dřeně a zabraňuje cirkulaci myelomových plazmocytů v periferní krvi, stejně jako jejich migraci extramedulárně (9, 18). Dále se u PCL vyskytuje snížená exprese antigenu CD106 a aktivované CD29 (31), snížená exprese povrchových molekul HLA-1 a CD40 na rozdíl od MGUS. Rovněž byla prokázána vyšší exprese CD54 v porovnání s adhezivními molekulami CD11a, CD18 a CD11b (32). Vysoká exprese VLA-4 u PCL je potřebná pro invazivnost leukemických buněk, kdy je nutná k navázání na svoji ligandu na povrchu kapilární stěny, čímž snáze dojde k extravazaci leukemických buněk do extravaskulárního prostoru (33, 34). Rovněž cytokiny jsou zapojené do proliferace PCL, zejména IL-6. Primární a sekundární PCL vykazují spontánní buněčný růst v kulturách, který se zrychlí při stimulaci exogenním IL-6 (35). Autokrinní produkce IL-6 spouštěná interferonem alfa (INF-alfa) byla prokázána u nemocného léčeného INF-alfa, u kterého se rozvinul obraz PCL, což potvrzuje potenciál cytokinové sítě v patogenezi této nemoci (36). Další důležitou molekulou podílející se na extramedulárním šíření je VEGF (vascular endothelial growth factor), který aktivací signální dráhy PI3K a MEK/ERK ovlivňuje migraci, proliferaci a přežívání myelomových buněk. Studie potvrzují, že tento faktor hraje zásadní roli v angiogenezi kostní dřeně, která je nezbytná pro šíření nádorových buněk. Dalším faktorem, jenž produkují plazmatické buňky je IGF1 (Insulin-like growth factor 1), který rovněž podporuje proliferaci a migraci nádorových buněk (37). Konečně byla diskutována souvislost s virovými infekcemi jako HIV, herpes virus 8 a virus hepatitidy C – nicméně s opačnými důsledky (38, 39). Další bádání nad patogenetickou rolí povrchových molekul, které mohou být zodpovědné za extramedulární šíření, je nutné.
Kritéria pro léčebnou odpověď
Neexistují žádná specifická kritéria léčebné odpovědi u PCL. Je obvyklé, že kritéria používaná při hodnocení léčby u MM jsou používaná i u PCL. Vzhledem k leukemické povaze onemocnění je důležité důsledné zhodnocení plazmocytů. Ty by měly být hodnoceny v periferní krvi, ale i v kostní dřeni jak morfologicky, tak pomocí průtokové cytometrie, stejně tak by měly být stanoveny hladiny volných lehkých řetězců. V práci Fernandez et al. bylo navrženo experty International Myeloma Working Party, aby při hodnocení léčebné odpovědi u primární PCL byla používána kombinovaná kritéria jak u akutní leukemie, tak u MM (5, 40) – tabulka 2. V případě PCL by měl být počet plazmocytů v kostní dřeni pod 5 % k hodnocení parciální remise, zatímco pro kompletní remisi je nezbytné vymizení plazmocytů v kostní dřeni při konvenčním morfologickém hodnocení. Pro hodnocení „striktní“ kompletní remise (stringent CR) by měl být požadován negativní výsledek vyšetření kostní dřeně a periferní krve pomocí průtokové cytometrie. Vysoká přítomnost extramedulárního postižení při PCL ospravedlňuje použití zobrazovacích technik, jako je magnetická rezonance (MRI) a zejména FDG pozitronová emisní tomografie či PET/CT.

PROGNÓZA NEMOCNÝCH A DLOUHODOBÉ PŘEŽITÍ
Prognóza je u nemocných s PCL vždy velmi nepříznivá. Přežití nemocných v publikovaných malých souborech bylo krátké (7–13 měsíců), přežití 5 let méně než 10% (41, 42). Medián přežití 231 nemocných v největší epidemiologické studii byl pouze 4 měsíce (43). Při použití autologní transplantace kostní dřeně bylo celkové přežití delší než 3 roky pozorované u 64 % nemocných (44). Je nutné zdůraznit, že zlepšení přežití pozorované u MM v poslední dekádě není bohužel zjistitelné u nemocných s PCL (43). Tyto znepokojující výsledky přežití u PCL jsou způsobeny dvěma základními faktory: agresivním projevem s komplikacemi způsobujícími smrt během prvních několika měsíců od diagnózy a absencí efektivní léčby, kterou by bylo možné dosáhnout trvalých výsledků. Časná mortalita je obrazem agresivity této nemoci. Ve francouzské studii zemřelo 28 % (11/40) nemocných během prvního měsíce od diagnózy (45). Sekundární PCL je většinou terminálním stavem s mediá-nem celkového přežití v délce jednoho měsíce (7).
LÉČEBNÉ MOŽNOSTI
Diagnózu PCL je nutné stanovit včas a okamžitě zahájit léčbu. Cílem indukční léčby by měla být co nejrychlejší kontrola nemoci tak, aby se maximálně snížilo riziko časné smrti. Je nezbytné říct, že léčebná doporučení jsou založena pouze na nekompletních údajích a na názorech odborníků. Neexistují randomizované klinické studie u PCL, neboť jde o raritní onemocnění.
Vstupní indukce by měla být pokud možno intenzivní. Mezi nejvhodnější patří režimy jako HyperCVAD (hyper-frakcionovaný cyklofosfamid, vinkristin, doxorubicin, dexametazon) (46) nebo PACE a kombinace založené na bortezomibu (VTD-PACE (bortezomib, thalidomid, dexametazon-cisplatina, doxorubicin, cyklofosfamid a etoposid), HyperCVAD-VD). Použití bortezomibu zlepšuje výsledky a tato látka se stala základním stavebním kamenem v léčbě PCL (47). Strategie vedoucí ke zlepšení dlouhodobého přežití jsou zahrnuty vždy, pokud není kontraindikace i na vysokodávkovanou chemoterapii s autologní transplantací hematopoetických kmenových buněk. Role konsolidační a udržovací léčby ještě není zhodnocena. Stejně tak zbývá definovat účinek jak tandemové autologní transplantace, tak alogenní transplantace. U nemocných mladších 50 let s vhodným dárcem (příbuzný nebo nepříbuzný) by měla být alogenní transplantace s myeloablativním režimem zvažovanou variantou léčby. Jako doporučený postup je na základě limitovaných zkušeností ve světě považován postup s využitím tandemové autologní transplantace následované alogenní transplantací s použitím režimu s redukovanou intenzitou (schéma 1A). U nemocných, kteří nejsou schopni podstoupit transplantaci, je možno použít režim na bázi bortezomibu, aby bylo dosaženo co nejrychlejší odpovědi – schéma 1B (47).

Léčba sekundární PCL nebo relapsu primární PCL závisí na léčebné odpovědi na předchozí léčbu a délce klidového období. Řada nemocných může mít přínos z režimů na bázi bortezomibu nebo z intenzivních režimů (např. HyperCVAD nebo Dexamethason – PACE) nebo mohou být zařazeni do časné fáze klinických studií (schéma 1C). Jinak je jedinou reálnou možností co nejkvalitnější podpůrná terapie.
Vysokodávkovaná myeloablativní chemoterapie s podporou autologních kmenových krvetvorných buněk (v textu zjednodušeně uváděno „autologní transplantace“)
Vzhledem k velmi špatné prognóze této plazmocytární dyskrazie by mělo být přistoupeno k intenzifikované vysokodávkované chemoterapii následované autologní transplantací, pokud to dovoluje věk a klinický stav nemocných. Ve studiích Mayo Clinic měli nemocní, kteří podstoupili autologní transplantaci, delší celkové přežití v porovnání s těmi, kteří byli léčeni pouze chemoterapií (průměr: 34 vs. 11 měsíců). Tyto výsledky jsou nepochybně do jisté míry zkresleny selekčními kritérii ve prospěch transplantační skupiny (3).
Nejrozsáhlejší studie zahrnující transplantaci byla provedena EBMT – European Group for Blood and Marrow Transplantation (48). Studie retrospektivně analyzovala data 272 nemocných s primární PCL ve srovnání s 20 844 nemocnými s MM. Z této studie vyplývá, že pPCL se svým klinickým chováním velmi podobá vysoce rizikovému MM, který dosahuje dobré léčebné odpovědi na vstupní indukční léčbu, nicméně krátké trvání léčebné odpovědi je následováno rychlým relapsem. Přestože nemocní s primární PCL dosáhli signifikantně častěji kompletní remise po autologní transplantaci (ve 100. dnu po transplantaci 41,2 % u primární PCL vs. 28,2 % u MM), průměrné přežití bez progrese bylo pouze 14,3 měsíce u primární PCL oproti 27,4 měsíce u MM. To se promítlo i do statisticky významně kratšího celkového přežití (medián 25,7 vs. 62,3 měsíců). Třebaže bylo u některých nemocných s primární PCL dosaženo relativně dobré odpovědi na úvodní léčbu a podstoupili intenzifikovaný režim s autologní transplantací, tak se nedočkali delšího přežití. Je tedy zjevné, že je třeba zkoušet jiné léčebné přístupy ke zlepšení prognózy nemocných s PCL, jako je použití nových látek při indukci, konsolidaci či udržovací léčbě nebo alogenní přístup. Na druhou stranu je autologní transplantace pravděpodobně v současné době nezbytná pro zvýšení šance na alespoň střednědobé přežití. CIBMTR (Center for International Blood and Marrow Transplant Research) zhodnotila střednědobé výsledky 97 nemocných s primární PCL, kteří podstoupili autologní transplantaci. Doba do relapsu byla ve třech letech sledování 34 %, celkové přežití 62 %. Tyto výsledky poprvé ukazují přežití delší než 3 roky u vybraných nemocných (44).
Role tandemové autologní transplantace nebyla zatím úplně zhodnocena a představuje příležitost k prohloubení a prodloužení remise. Jiné strategie ke konsolidaci po první autologní transplantaci zahrnuje režimy VTD, RD, VRD, které zasluhují další prospektivní zhodnocení. Přidání dlouhotrvající udržovací terapie s lenalidomidem nebo moderní kombinace založené na lenalidomidu nabízí další potenciální strategii k prodloužení trvání remise. Významný přínos na přežití bez progrese, zveřejněný ve dvou studiích s udržovací léčbou lenalidomidem po provedení autologní transplantace naznačuje, že by tato léčebná strategie mohla být zajímavou možností ve výzkumu PCL (48, 49). Relaps se může objevit záhy po provedení autologní transplantace, a proto je nutné zvážit velmi časné zahájení udržovací terapie, a to ihned, jak je zřejmé stabilní přihojení štěpu.
Alogenní transplantace kostní dřeně
Retrospektivní výsledky u 147 nemocných s primární PCL čerpající ze záznamů CIBMTR ukazují, že 39 % (19/50) nemocných bylo naživu ještě 3 roky po provedení alogenní transplantace kostní dřeně (44). Část nemocných dostala moderní látky (bortezomib, thalidomid, lenalidomid) jako součást indukčního režimu. Na progresi onemocnění zemřelo 22 % nemocných ve skupině, která podstoupila alogenní transplantaci, zatímco ve skupině, která podstoupila autologní transplantaci, to byl téměř čtyřnásobek (85 %). EBMT nedávno popsala zkušenost s 85 nemocnými, kteří podstoupili alogenní transplantaci kostní dřeně v porovnání se 411 nemocnými, kteří podstoupili autologní transplantaci. Křivky přežití bez progrese s myeloablativní a RIC (Reduced Intensity Conditioning) přípravou k alogenní transplantaci křížily křivku autologních transplantací mezi 2. a 4. rokem, nicméně celkové přežití bylo stejné – 5 let. Jak můžeme vidět z předešlých zkušeností CIMBTR s alogenními transplantacemi, vždy je přítomna vysoká časná úmrtnost, ale současně existuje zřejmé plató přežití někde okolo 20 % (50). Alogenní přístup je jistou nadějí pro velmi limitovaný počet nemocných s primární PCL, přihlédneme-li k věku nemocných, dostupností vhodného dárce, časné transplantační úmrtnosti a celkovým výsledkům. U sekundární PCL není zpravidla alogenní transplantace vůbec indikovaná.
NOVÉ LÉKY
Konvenční léčba běžnými dávkami u MM je zpravidla neúčinná. Role thalidomidu je zanedbatelná (51). Byla u něj popsána poměrně vážná kardiální a pulmonární toxicita (52, 53). Lenalidomidem bylo dosaženo pouze přechodné léčebné odpovědi (54, 55). Ve fázi II prospektivní multicentrické studie, do které bylo zařazeno 23 nemocných s primární PCL, byla hodnocena v první léčebné linii kombinace lenalidomid a dexametazon (56). Nemocní užívali lenalidomid 25 mg – den 1.–21. a jednou týdně 40 mg dexametazonu v 28denním cyklu. Pokud byli nemocní schopni, tak podstoupili autologní transplantaci a pokračovali v dlouhodobém podávání primární terapie. Vstupní léčebná odpověď byla dosažena u 61 % (14/23) nemocných. V 15. měsíci (medián) sledování bylo celkové přežití a přežití bez progrese 65,2 % (15/23), respektive 52,1 % (12/23).
Bortezomib
Režimy založené na bortezomibu se staly základním terapeutickým kamenem v léčbě PCL. Tento inhibitor proteazomu ukázal klinickou aktivitu jak u primární, tak u sekundární PCL (57). Italská skupina uveřejnila výsledky retrospektivní analýzy 12 nemocných s relapsem primární PCL a sekundární PCL léčených kombinací založené na bortezomibu (58). Bylo dosaženo léčebné odpovědi u 92 % (11/12) nemocných včetně dvou kompletních remisí. Průměrné přežití bez progrese a celkové přežití po bortezomibu bylo 8, respektive 12 měsíců. Stejná skupina popsala podobně vysokou odpověď u nemocných s nově diagnostikovanou primární PCL léčených bortezomibem v různých kombinovaných režimech (VD (bortezomib, dexametazon), n = 3, VTD (bortezomib, thalidomid, dexametazon), n = 2, PAD (bortezomib, doxorubicin, dexametazon), n = 6, MPV (melfalan, prednizon, bortezomib), n = 1). Léčebná odpověď byla pozorovaná u 79 % (23/29) nemocných včetně 28 % (8/29) s kompletní remisí. Celkem 83 % (24/29) nemocných podstoupilo autologní transplantaci. I přes krátké sledování byla ztráta nemocných v indukční fázi poměrně nízká (17 %, 5/29) s ohledem na rizikovost onemocnění (47). Existují další studie převážně na malém počtu nemocných, které dokazují přínos bortezomibu v léčbě tohoto vážného onemocnění (59), stejně tak jsou účinné kombinace bortezomibu s dexametazonem a melfalanem či doxorubicinem (60, 61).
ZÁVĚR
Plazmocelulární leukemie je velmi vzácná agresivní forma monoklonální gamapatie. V diagnostice PCL stojí na prvním místě morfologické zhodnocení nátěrů periferní krve s nálezem plazmocytů, diagnostická kritéria jsou v tuto chvíli celosvětově diskutována a v dohledné době mohou být upravena. Podobně se diskutuje o léčebných kritériích. Návrhem je používat kombinované hodnocení léčebné odpovědi. V léčbě PCL se používají konvenční léčebné režimy v kombinaci s moderními látkami, zejména s bortezomibem. Indukční léčba se zahajuje ihned po stanovení diagnózy, strategie zlepšující dlouhodobé přežití zahrnují vysokodávkovanou chemoterapii s následnou autologní transplantací kmenových buněk, u mladších nemocných vzhledem k agresivitě nemoci připadá v úvahu i alogenní transplantace. Léčba sekundární PCL a relapsu primární PCL je velmi obtížná a obecně neúspěšná.
Poděkování: Autoři děkují paní MUDr. Janě Zuchnické (Klinika hematoonkologie, Fakultní nemocnice Ostrava) za poskytnutí morfologických obrázků plazmocelulární leukemie, Mgr. Henrietě Grešlíkové a doc. RNDr. Petrovi Kuglikovi, CSc. (Babákova myelomová skupina při Ústavu patologické fyziologie LF MU, Brno) za poskytnutí CGH array PCL a dále paní Ing. Lence Mršťákové (Oddělení pro vědu a výzkum, Lékařská fakulta, Ostravská univerzita) za administrativní podporu při úpravě publikace, tvorbě tabulek a formátování textu a literatury. Tato práce vznikla za přispění Institucionální podpory Ministerstva zdravotnictví ČR č. 1 RVO-FNOs/2012. Projekt byl rovněž financován v rámci Institucionálního rozvojového plánu OU v Ostravě v roce 2012. Finanční prostředky přiděluje MŠMT.
Podíl autorů na přípravě rukopisu
T. Jelínek – napsání rukopisu
H. Plonková – podíl na sepsání rukopisu, příprava podpůrné grafické dokumentace, revize dokumentů
R. Hájek – návrh tématu, osnovy práce, finální kontrola a revize rukopisu
Doručeno do redakce: 10. 4. 2013
Přijato po recenzi: 10. 6. 2013
Prof. MUDr. Roman Hájek, CSc.
Klinika hematoonkologie FN Ostrava
17. listopadu 1790
708 52 Ostrava
e-mail: roman.hajek@fno.cz
Zdroje
1. Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010; 116 : 3724-3734.
2. Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R. Primary plasma cell leukaemia. Br J Haematol 1994; 88 : 754-759.
3. Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008; 22 : 1044-1052.
4. Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002. Cancer Causes Control 2008; 19 : 379-390.
5. Fernandez C, Kyle RA, Durie BGM, Lidwig H, Usmani S. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group, Leukemia 2013; 27 : 780-791.
6. Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med 1974; 133 : 813-818.
7. Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med 1987; 83 : 1062-1068.
8. Bladé J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am 1999; 13 : 1259-1272.
9. García-Sanz R, Orfao A, González M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood 1999; 93 : 1032-1037.
10. Pagano L, Valentini CG, De Stefano V, et al. Primary plasma cell leukemia: a retrospective multicenter study of 73 patients. Ann Oncol 2011; 22 : 1628-1635.
11. Kyle RA. Multiple myeloma: review of 869 cases. Mayo Clin Proc 1975; 50 : 29-40.
12. Dimopoulos MA, Barlogie B, Smith TL, Alexanian R. High serum lactate dehydrogenase level as a marker for drug resistance and short survival in multiple myeloma. Ann Intern Med 1991; 115 : 931-935.
13. Kumar S, Rajkumar SV, Kyle RA, et al. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J Clin Oncol 2005; 23 : 5668-5674.
14. Nowakowski GS, Witzig TE, Dingli D, et al. Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood 2005; 106 : 2276-2279.
15. Shtalrid M, Shvidel L, Vorst E. Polyclonal reactive peripheral blood plasmacytosis mimicking plasma cell leukemia in a patient with Staphylococcal sepsis. Leuk Lymphoma 2003; 44 : 379-380.
16. Touzeau C, Pellat-Deceunynck C, Gastinne T, et al. Reactive plasmacytoses can mimick plasma cell leukemia: therapeutical implications. Leuk Lymphoma 2007; 48 : 207-208.
17. Al-Sahmani M, Trnavska I, Sevcikova S, et al. Prognostic significance of morphological assessment of plasma cells in multiple myeloma, Neoplasma 2011, 58 : 554-560.
18. Pellat-Deceunynck C, Barillé S, Jego G, et al. The absence of CD56 (NCAM) on malignant plasma cells is a hallmark of plasma cell leukemia and of a special subset of multiple myeloma. Leukemia 1998; 12 : 1977-1982.
19. Guikema JE, Vellenga E, Abdulahad WH, Hovenga S, Bos NA. CD27triggering on primary plasma cell leukaemia cells has anti--apoptotic effects involving mitogen activated protein kinases. Br J Haematol 2004; 124 : 299-308.
20. Guikema JE, Hovenga S, Vellenga E, et al. CD27 is heterogeneously expressed in multiple myeloma: low CD27 expression in patients with high-risk disease. Br J Haematol 2003; 121 : 36-43.
21. Walters M, Olteanu H, Van Tuinen P, Kroft SH. CD23 expression in plasma cell myeloma is specific for abnormalities of chromosome 11, and is associated with primary plasma cell leukaemia in this cytogenetic sub-group. Br J Haematol 2010; 149 : 292-293.
22. van de Donk NW, Lokhorst HM, Anderson KC, Richardson PG. How I treat plasma cell leukemia, Blood 2012; 120 : 2376-2389.
23. Chang H, Yeung J, Xu W, Ning Y, Patterson B. Significant increase of CKS1B amplification from monoclonal gammopathy of undetermined significance to multiple myeloma and plasma cell leukaemia as demonstrated by interphase fluorescence in situ hybridisation. Br J Haematol 2006; 134 : 613-615.
24. Bezieau S, Devilder MC, Avet-Loiseau H, et al. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum Mutat 2001; 18 : 212-224.
25. Urashima M, Teoh G, Ogata A, et al. Characterization of p16(INK4A) expression in multiple myeloma and plasma cell leukemia. Clin Cancer Res 1997; 3 : 2173-2179.
26. Mateos MV, Garcia-Sanz R, López-Pérez R, et al. p16/INK4a gene inactivation by hypermethylation is associated with aggressive variants of monoclonal gammopathies. Hematol J 2001; 2 : 146-149.
27. Bollati V, Fabris S, Pegoraro V, et al. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009; 30 : 1330-1335.
28. Usmani SZ, Nair B, Qu P, et al. Primary plasma cell leukemia: clinical and laboratory presentation, gene-expression profiling, and clinical outcome with Total Therapy protocols. Leukemia, 2012; 26 : 2398-2405.
29. Egan JB, Shi CX, Tembe W, et al. Whole genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution and clonal tides. Blood 2012; 120 : 1060-1066.
30. Mitsiades CS, McMillin DW, Klippel S, et al. The role of bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am 2007; 21 : 1007-1034.
31. Luque R, García-Trujillo JA, Cámara C, et al. CD106 and activated-CD29 are expressed on myelomatous bone marrow plasma cells and their downregulation is associated with tumour progression. Br J Haematol 2002; 119 : 70-78.
32. Kraj M, Kopeć-Szlęzak J, Pogłód R, Kruk B. Flow cytometric immunophenotypic characteristics of 36 cases of plasma cell leukemia. Leuk Res 2011; 35 : 169-76.
33. Pérez-Andrés M, Almeida J, Martín-Ayuso M, et al. Clonal plasma cells from monoclonal gammopathy of undetermined significance, multiple myeloma and plasma cell leukemia show different expression profiles of molecules involved in the interaction with the immunological bone marrow microenvironment. Leukemia 2005; 19 : 449-455.
34. Vande Broek I, Vanderkerken K, Van Camp B, Van Riet. Extravasation and homing mechanisms in multiple myeloma. Clin Exp Metastasis 2008; 25 : 325-334.
35. Zhang XG, Bataille R, Widjenes J, Klein B. Interleukin-6 dependence of advanced malignant plasma cell dyscrasias. Cancer 1992; 69 : 1373-1376.
36. Bladé J, López-Guillermo A, Tassies D, Montserrat E, Rozman C. Development of aggressive plasma cell leukaemia under interferon--alpha therapy. Br J Haematol 1991; 79 : 523-525.
37. Fišerová B, Kubiczková L, Sevčíková S, Hájek R. Implication of bone marrow microenvironment in pathogenesis of multiple myeloma. Klin Onkol 2012; 25 : 234-240.
38. Duprez R, Lacoste V, Hermouet S, et al. Plasma-cell leukemia and human herpesvirus 8 infection. Leukemia 2004; 18 : 1903-1904.
39. Hermouet S, Corre I, Gassin M, Bigot-Corbel E, Sutton CA, Casey JW. Hepatitis C virus, human herpesvirus 8, and the development of plasma-cell leukemia. N Engl J Med 2003; 348 : 178-179.
40. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood 2010; 115 : 453-474.
41. Colović M, Janković G, Suvajdzić N, Milić N, Dordević V, Janković S. Thirty patients with primary plasma cell leukemia: a single center experience. Med Oncol 2008; 25 : 154-160.
42. Peijing Q, Yan X, Yafei W. A retrospective analysis of thirty-one cases of plasma cell leukemia from a single center in China. Acta Haematol 2009; 121 : 47-51.
43. Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D. Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer 2009; 115 : 5734-5739.
44. Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia 2012; 26 : 1091-1097.
45. Avet-Loiseau H, Daviet A, Brigaudeau C, et al. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myélome and the Groupe Français de Cytogénétique Hématologique. Blood 2001; 97 : 822-825.
46. Saccaro S, Fonseca R, Veillon DM, et al. Primary plasma cell leukemia: report of 17 new cases treated with autologous or allogeneic stem-cell transplantation and review of the literature. Am J Hematol 2005; 78 : 288-294.
47. D’Arena G, Valentini CG, Pietrantuono G, et al. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol 2012; 23 : 1499-1502.
48. Drake MB, Iacobelli S, van Biezen A, et al. European Group for Blood and Marrow Transplantation and the European Leukemia Net. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica 2010; 95 : 804-809.
49. Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366 : 1782-1791.
50. Morris C, Iacobelli S, Gahrton G, et al. Has allogeneic transplantation a role in the management of plasma cell leukaemia? A study on behalf of the Myeloma Subcomittee of the Chronic Leukaemia Working Party of the EBMT. Blood (ASH Annual Meeting Abstracts) 2011; 118: Abstract 2008.
51. Petrucci MT, Martini V, Levi A, et al. Thalidomide does not modify the prognosis of plasma cell leukemia patients: experience of a single center. Leuk Lymphoma 2007; 48 : 180-182.
52. Pretz J, Medeiros BC. Thalidomide-induced pneumonitis in a patient with plasma cell leukemia: no recurrence with subsequent lenalidomide therapy. Am J Hematol 2009; 84 : 698-699.
53. Ballanti S, Mastrodicasa E, Bolli N, et al. Sustained ventricular tachycardia in a thalidomide-treated patient with primary plasma-cell leukemia. Nat Clin Pract Oncol 2007; 4 : 722-725.
54. Musto P, Pietrantuono G, Guariglia R, et al. Salvage therapy with lenalidomide and dexamethasone in relapsed primary plasma cell leukemia. Leuk Res 2008; 32 : 1637-1638.
55. Olivieri A, Attolico I, Cimminiello M, Discepoli G, Cifarelli RA. Lenalidomide can induce graft versus leukemia effect in primary plasma cell leukemia: a case report. Leuk Res 2009; 33 : 191-193.
56. Musto P, D’Auria F, Petrucci MT, et al. Final results of a phase II study evaluating lenalidomide in combination with low dose dexamethasone as first line therapy for primary plasma cell leukemia. Blood (ASH Annual Meeting Abstracts), 2011; 118: Abstract 2925.
57. Esparis-Ogando A, Alegre A, Aguado B, et al. Bortezomib is an effi-cient agent in plasma cell leukemias. Int J Cancer 2005; 114 : 665-667.
58. Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer 2007; 109 : 2285-2290.
59. Lebovic D, Zhang L, Alsina M, et al. Clinical outcomes of patients with plasma cell leukemia in the era of novel therapies and hematopoietic stem cell transplantation strategies: a singleinstitution experience. Clin Lymphoma Myeloma Leuk 2011; 11 : 507-511.
60. Libby E, Candelaria-Quintana D, Moualla H, Abdul-Jaleel M, Rabinowitz I. Durable complete remission of primary plasma cell leukemia with the bortezomib plus melphalan and prednisone (VMP) regimen. Am J Hematol 2010; 85 : 733-734.
61. Al-Nawakil C, Tamburini J, Bardet V, et al. Borderline, doxorubicin and dexamethasone association is an effective option for plasma cell leukemia induction therapy. Leuk Lymphoma 2008; 49 : 2012-2014.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2013 Číslo 3
- Dentální extrakce u hemofiliků
- Sport nemění u zdravých mužů skóre HJHS
- Rizikové faktory pro rozvoj inhibitorů u dětí s hemofilií – výsledky kohortové studie
- Kazuistika: Neobvyklá příčina prodlouženého APPT u SLE – získaná hemofilie a lupus antikoagulans
- Ekonomický význam léčby krvácení u pacientů s inhibitorem pomocí rFVIIa
Najčítanejšie v tomto čísle
- Současná léčba myelofibrózy na základě rizikové stratifikace pacientů
- Lymfomy gastrointestinálního traktu – klinicko-patologický přehled
- Význam stanovení sérových hladin volných lehkých řetězců imunoglobulinu u AL amyloidózy
- Plazmocelulární leukemie