
Vzácný pyrofosfátový močový konkrement u pětiletého chlapce s kongenitální hypofosfatázií
Rare pyrophosphate renal stone in 5 years old boy with congenital hypophosphatasia
Objectives: Identification of unusual renal pyrophosphate stone in 5 years old patient with congenital hypophosphatasia.
Design: Case report and evaluation of laboratory results.
Settings: Department of Medical Chemistry and Clinical Biochemistry, University Hospital Motol, Charles University, Second Faculty of Medicine, Department of Paediatrics, University Hospital Motol, Charles University, Second Faculty of Medicine, V Úvalu 84, 150 06 Prague 5 (Czech Republic).
Material and methods: Case report focusing on determination of urinary stone composition by polarized light microscopy and infra-red spectrometry.
Results: In 5 years old patient with effectively treated congenital hypophosphatasia by recombinant alkaline phosphatase (the serum value of which was ten times the upper limit of the reference range), rare pyrophosphate renal stone with addition of sulphate was demonstrated. The formation of the stone occurred in the period when the patient´s urine was thera-peutically alkalized (due to the history of urate stones) and the patient used prophylactically sulfonamides prior to planned lithotripsy.
Conclusion: Despite the high serum alkaline phosphatase concentration, the function of which is the conversion of pyrophosphate to phosphate, pyrophosphate renal stone was identified in a patient with congenital hypophosphatasia. The sulphate component of the stone is probably due to the prophylactic administration of sulfonamides.
Keywords:
renal stone – pyrophosphate – sulphonamides – congenital hypophosphatasia – infra-red spectrometry
Autoři:
M. Polák 1; K. Kotaška 1
![]() ; L. Plachý 2; R. Průša 1; M. Fořtová 1
; L. Plachý 2; R. Průša 1; M. Fořtová 1
Působiště autorů:
Ústav lékařské chemie a klinické biochemie 2. LF UK a FN Motol, Praha
1; Pediatrická klinika 2. LF UK a FN Motol, Praha
2
Vyšlo v časopise:
Klin. Biochem. Metab., 27, 2019, No. 2, p. 90-95
Souhrn
Cíl: Popsat nález neobvyklého močového konkrementu u pětiletého pacienta s kongenitální hypofosfatázií.
Typ studie: Kazuistika se zhodnocením laboratorních nálezů.
Název a sídlo pracoviště: Ústav lékařské chemie a klinické biochemie 2. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice Motol, Pediatrická klinika 2. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice Motol, V Úvalu 84, 150 06 Praha 5
Materiál a metody: Popis kazuistiky se zaměřením se především na analýzy močových konkrementů pomocí polarizační mikroskopie a infračervené spektrometrie.
Výsledky: U pětiletého pacienta s kongenitální hypofosfatázií efektivně léčeného rekombinantní alkalickou fosfatázou (jejíž hodnota v séru představovala desetinásobek horní hranice referenčního rozmezí) byl prokázán raritní pyrofosfátový močový konkrement s příměsí sulfátu. K jeho tvorbě došlo v období, kdy byla u pacienta terapeuticky alkalizována moč (jejímž důvodem byla metafylaxe urátové urolitiázy), a pacient užíval profylakticky sulfonamidy před plánovanou litotrypsí.
Závěr: Ačkoliv sérová koncentrace alkalické fosfatázy, jejíž funkcí je přeměna pyrofosfátu na fosfát, byla vysoká, byl u pa-cienta s kongenitální hypofosfatázií identifikován pyrofosfátový močový konkrement. Sulfátová složka konkrementu vznikla pravděpodobně na podkladě profylaktického podávání sulfonamidů.
Klíčová slova:
močový konkrement – pyrofosfát – sulfonamidy – vrozená hypofosfatázie – infračervená spektrometrie
Úvod
Urolitiáza je považována za civilizační onemocnění. Postihuje nejčastěji pacienty mezi 20. až 60. rokem života, s incidencí 6 až 7 % u žen a 11 až 15 % u mužů. V poslední dekádě je pozorován nárůst prevalence vzniku onemocnění u dětské populace, přibližně 10 % ze všech případů urolitiázy je diagnostikováno právě u dětí [1].
Rizikové faktory vedoucí k rozvoji urolitiázy jsou dobře známy. Jedná se zejména o zvýšený obsah litogenních látek (např. kalciové a fosfátové ionty, oxaláty, sodné ionty a kyselina močová), snížené množství látek působících inhibičně (např. citráty, hořečnaté ionty, Tamm-Horsfallův mukoprotein, pyrofosfát), nižší příjem tekutin s diurézou pod 1500 mL moče, změna pH moče (např. renální tubulární acidóza, bakteriální infekce), stravování (např. vysoký příjem živočišných bílkovin, solí, alkoholu, cukrů a fosfátů) a vrozené metabolické poruchy (např. poruchy metabolizmu oxalátů, purinů, cystinu) [2, 3].
V této kazuistice popisujeme nález raritního pyrofosfátového konkrementu u pacienta s kongenitální hypofosfatázií.
Hypofosfatázie (HPP) je vzácné onemocnění charakterizované dominujícím postižením skeletu a mnoha systémovými projevy vzniklými v důsledku snížené sérové koncentrace tkáňově nespecifické alkalické fosfatázy [4]. Je způsobena mutací ALPL (liver/bone/kidney alkaline phosphatase) genu, jenž kóduje tkáňově nespecifickou alkalickou fosfatázu (TNALP). Gen (70 kb) je lokalizován na 1. chromozomu (1p36.12) a obsahuje 12 exonů. V současné době je známo 388 mutací tohoto genu [5, 6]. TNALP je fosfomonoesteráza a je aktivní pouze ve formě dimeru. Její základní biochemickou funkcí je odštěpování fosfátů z různých molekul pomocí hydrolýzy. Štěpí tyto substráty: pyridoxal-5´-fosfát (aktivní metabolit vitaminu B6), fosfoetanolamin a anorganický pyrofosfát. Kvůli defektní TNALP se tyto substráty hromadí v krvi a v moči. TNALP se podílí na mineralizaci kostí a zubů [7], kdy konverzí pyrofosfátu na fosfát umožňuje tvorbu hydroxyapatitu [8]. Pyrofosfát je po chemické stránce difosfát (P2O74-), aniont kyseliny difosforečné, vznikající např. při štěpení ATP ligázami [9]. Do krve se dostává především jako metabolit jaterních buněk [10], jeho koncentrace v plazmě je 1 až 6 μmol/L [11]. V plazmě je rychle hydrolyzován a zbytek filtrován glomeruly. V ledvinách dochází k další hydrolýze a pouze méně než 5 % plazmatického pyrofosfátu se dostává do definitivní moči [12]. V proximálních tubulech ledvin je exprimována TNALP, v jejich pars recta také střevní izoenzym ALP [13]. Exprese v proximálních tubulech zajišťuje časnou hydrolýzu pyrofosfátů v primární moči. Proximální i distální části nefronu jsou schopny pyrofosfát také samy generovat z ATP nebo z dalších nukleotid trifosfátů [3].
Defosforylací pyridoxal-5´-fosfátu usnadňuje TNALP jeho transport přes hematoencefalickou bariéru, čímž mu umožní vykonávat funkci neurotransmiteru v mozkových buňkách. Někteří pacienti s HPP se proto manifestují křečemi, které jsou pyridoxin senzitivní. V ledvinách TNALP stimuluje exkreci fosfátů, pro pacienty s HPP bývá typická hyperfosfatémie [4].
Hypofosfatázie se dědí nejčastěji autozomálně recesivně (těžší formy) nebo autozomálně dominantně (lehčí formy), může se jednat i o mutace de novo. Incidence v populaci je v případě závažných forem v evropských populacích 1 : 300000 a v případě mírnějších forem 1 : 6300 [4].
Jedná se o klinicky heterogenní onemocnění, rozlišujeme pět základních forem: perinatální, infantilní, dětskou, adultní a odontohypofosfatázii. Perinatální forma je klinicky nejtěžší, projevuje se extrémní demineralizací skeletu a asfyxií. Bez substituční terapie alkalickou fosfatázou je letální většinou již v novorozeneckém období. Infantilní forma bývá diagnostikována do šesti měsíců věku. Bezprostřední postnatální adaptace nebývá alterována, v následujících týdnech se objevují nespecifické příznaky: neprospívání, svalová slabost, opožďování v psychomotorickém vývoji. Jsou nápadné deformity skeletu, rozvíjí se porucha růstu, dochází ke vzniku kraniosynostózy. K častým projevům patří tracheomalacie s tendencí k mnohdy velmi závažným respiračním infektům. Pacienti trpící dětskou formou jsou diagnostikováni po šesti měsících věku. Jedná se o velmi heterogenní skupinu, od mírných forem až po případy imitující infantilní formu. Nápadná je předčasná ztráta primární dentice a špatná kvalita permanentní dentice. Nálezy na skeletu připomínají rachitické změny, rozvíjí se porucha růstu, je opožděn vývoj motoriky. Adultní forma se typicky manifestuje ve středním věku, nejčastěji předčasnou ztrátou dentice nebo opakovanými, těžko se hojícími stresovými frakturami metatarzů nebo subtrochanterickou pseudofrakturou femuru. Odontohypofosfatázie je nejmírnější forma HPP projevující se výhradně stomatologickými příznaky [4].
Pokud v diferenciální diagnostice zjištěné metabolické kostní poruchy hypofosfatázii zvažujeme, bývá stanovení diagnózy jednoduché. Typické klinické symptomy provází velmi nízká koncentrace sérové ALP. V laboratorním nálezu můžeme vidět též hyperkalcémii, sekundární hypoparatyreózu (což odráží insuficietní mineralizaci skeletu), hyperfosfatémii a hyperkalciurii [4]. Právě hyperkalciurie může být příčinou nefrokalcinózy a poškození ledvin [14]. Pomocnými markery jsou fosfoetanolamin a pyridoxal 5´-fosfát, které se při nedostatku ALP v séru akumulují [4].
Materiál a metody
Anamnéza
Pětiletý chlapec byl přijat v listopadu 2018 na Pedia-trickou kliniku Fakultní nemocnice Motol k přešetření po 2. litotrypsi pro urolitiázu.
Chlapec se narodil z první gravidity v dokončeném 37. týdnu s porodní hmotností 2890 gramů a délkou 49 cm. Již prenatálně u něj bylo vyjádřeno podezření na zkrácení dlouhých kostí. Při narození vykazoval známky stigmatizace: měl extrémní velkou fontanelu s prostornějšími lebečními švy, otevřenou malou fontanelu, lebeční kosti byly palpačně měkké, končetiny kratší, klouby i kosti hypermobilní, na RTG porucha osifikace. Jeho poporodní adaptace byla komplikovaná v důsledku dyspnoe, hypotonie a cyanózy. Do dvou hodin po naro-zení se rozvinula respirační insuficience při nestabilitě hrudního koše s nutností ventilační podpory.
Pacient byl přeložen na Novorozenecké oddělení Fakultní nemocnice Motol. Laboratorně byla zjištěna extrémně nízká katalytická koncentrace alkalické fosfatázy (0,06 µkat/L, referenční rozmezí: 1,20 – 6,30 µkat/L), hyperkalcémie (3,0 mmol/L, referenční rozmezí: 1,75 až 2,87 mmol/L) při fyziologické koncentraci fosfátu (2,27 mmol/L, referenční rozmezí: 1,36 – 2,58 mmol/L) a PTH (1,86 pmol/L, referenční rozmezí: 1,30 – 7,60 pmol/L). Tyto nálezy společně s typickým fenotypem ukazují na těžkou perinatální formu hypofosfatázie. Diagnóza byla potvrzena molekulárně genetickým vyšetřením, které prokázalo mutaci v genu ALPL (1471 G > A, Gly491Arg) v homozygotní formě. Následná analýza DNA ukázala kompletní uniparentální maternální izodisomii celého 1. chromozomu, kde se nalézá alela pro gen ALPL [15, 16].
U pacienta byla nejprve zavedena konzervativní symptomatická terapie (vzhledem k nemožnosti kauzální léčby v České republice v době jeho narození): forsírovaná diuréza ke snížení kalcemie (v 1 měsíci věku zjištěna již normokalcemie 2,52 mmol/L při zvýšeném vylučování kalcia do moče 3,0 mmol/L, po korekci na kreatinin 5,0 mmol/mmol, referenční rozmezí podle [17]: <1,5 mmol/mmol), substituce vitaminu D (koncentrace kalcidiolu v jednom měsíci věku byla 48,7 nmol/L, referenční rozmezí 50,0 – 250,0 nmol/L) a jako prevence možného rozvoje křečí léčba pyridoxinem [15]. Od druhého měsíce věku je chlapci kauzálně podávána rekombinantní alkalická fosfatáza Asfotáza alfa (Strensiq, Alexion, 3x týdně subkutánně).
Do 15 měsíců věku byl pacient trvale na ventilační podpoře, průběh byl komplikován kardiální nestabilitou a laryngotracheobronchomalacií, byla provedena tracheostomie. V jednom roce věku byly zachyceny epileptické záchvaty charakteru absencí, na MRI nález posthypoxické malacie v oblasti bazálních ganglií, byla nasazena antiepileptika s dobrým efektem. Vzhledem k nálezu kraniosynostózy byla v 15 měsících věku provedena remodelace lbi. V 17 měsících prodělal kardiopulmonální resuscitaci pro srdeční zástavu při spontánní dekanylaci z tracheostomie. Ve dvou letech byla z MRI zjištěna pokročilá atrofizace mozku a posthypoxické změny. Neuropsychologické vyšetření ve čtyřech letech věku prokázalo, že motorický vývoj chlapce odpovídá I. trimenonu, navíc vykazuje spastickou kvadruparézu, mentální vývoj přelomu I. a II. trimenonu. Neurologické postižení má tendenci k progresi, v dalším vývoji se objevily např. fascikulace v obličeji, přežvykování, třes končetin. Chlapec opakovaně prodělal pneumonie s nutností oxygenoterapie, jejichž frekvence se mírně snižuje. Několikrát mu byl ošetřován kariézní chrup. Dostatečná výživa je zajištěna perkutánní endoskopickou gastrostomií.
Léčbu Asfotázou alfa pacient toleruje dobře, došlo k postupné kostní mineralizaci a ke zmírnění respiračních obtíží (ventiluje spontánně přes tracheostomii), je bez fraktur.
Urologické komplikace v anamnéze
Od dvou let věku se u pacienta opakovaly nefrolitiázy a ureterolitiázy, které byly řešeny zavedením stentů, nefrostomie a cystoskopickou extrakcí konkrementů. Jeden z konkrementů byl analyzován v naší laboratoři. Byl tvořen oranžovým jádrem obsahujícím 100 % uricitu a bílou povrchovou vrstvou obsahující 60 % uricitu a 40 % urátu amonného. Ve 4,5 letech pacient prodělal v důsledku urolitiázy urosepsi. Byla zachycena též významná proteinurie (23 g/L), ale celková bílkovina v séru ve fyziologickém rozmezí, v dalších odběrech již bez patologické proteinurie. Po antibiotickém zaléčení byla doporučena litotrypse emisní rázovou vlnou a do výkonu profylaxe kotrimoxazolem. Po litotrypsi však urolitiáza přetrvávala, proto bylo doporučeno její zopakování a ponechání zajištění kotrimoxazolem.
Laboratorní vyšetření
V průběhu hospitalizace bylo provedeno vyšetření biochemických parametrů v séru a v moči. V séru byla zjištěna vysoká koncentrace ALP 61,34 μkat/L (referenční rozmezí: 1,12 až 6,20 μkat/L), mírná hyperfosfatémie 1,98 mmol/L (referenční rozmezí: 1,16 až 1,90 mmol/L), normokalcémie 2,42 mmol/L (referenční rozmezí: 2,05 až 2,54 mmol/L), fyziologická koncentrace kalcidiolu 72,1 nmol/L (referenční rozmezí: 50 až 250 nmol/L) a kreatininu 21 μmol/L (referenční rozmezí: 4 až 40 μmol/L, odhadnutá glomerulární filtrace (Schwartz) 2,84 ml/s/1,73 m2). Hodnota PTH ze září 2018 byla fyziologická 1,60 pmol/L (referenční rozmezí: 1,30 až 7,60 pmol/L), anamnesticky byla zjištěna i hypoparatyreóza (1,00 pmol/L v prosinci 2017).
V moči byla zjištěna významná proteinurie 492,0 mg/L, po korekci na močový kreatinin 615,0 mg/mmol (referenční rozmezí: 0 až 22,7 mg/mmol) a albuminurie 127,7 mg/L, po korekci na močový kreatinin 159,6 mg/mmol (referenční rozmezí: < 3,3 mg/mmol). Hodnota močového kreatininu byla snížená 0,8 mmol/L (referenční rozmezí: 5,7 až 14,7 mmol/L). Kalciurie byla 0,54 mmol/L, korigovaná na močový kreatinin na horní hranici referenčního rozmezí 0,675 mmol/mmol (referenční rozmezí dle [17]: < 0,70 mmol/mmol), fosfaturie 3,2 mmol/L, po korekci na močový kreatinin 2,13 mmol/mmol, (referenční rozmezí dle [18]: 1,20 až 4,34 mmol/mmol).
Vyšetření moče chemicky a močového sedimentu ukázalo následující nálezy: specifická hmotnost moče 1,005 kg/L (referenční rozmezí: 1,018 až 1,025 kg/L), pH 7,0 (referenční rozmezí: 5,0 až 7,0), leukocyty 706 částic/µl (referenční rozmezí: 0 až 15 částic/µl), erytrocyty 15 částic/µl (referenční rozmezí: 0 až 10 částic/µl), bakteriurie.
Vyšetření konkrementu
Pacient po provedení 2. litotrypse vyloučil v odstupu čtyř týdnů celkem devět konkrementů. Konkrementy byly postupně odeslány do naší laboratoře k chemickému rozboru a byly analyzovány pomocí polarizační mikroskopie (Jenapol, Carl Zeiss Jenna, Německo) a infračervené spektrometrie (Nicolet iS10, Thermo Fischer Scientific, USA).
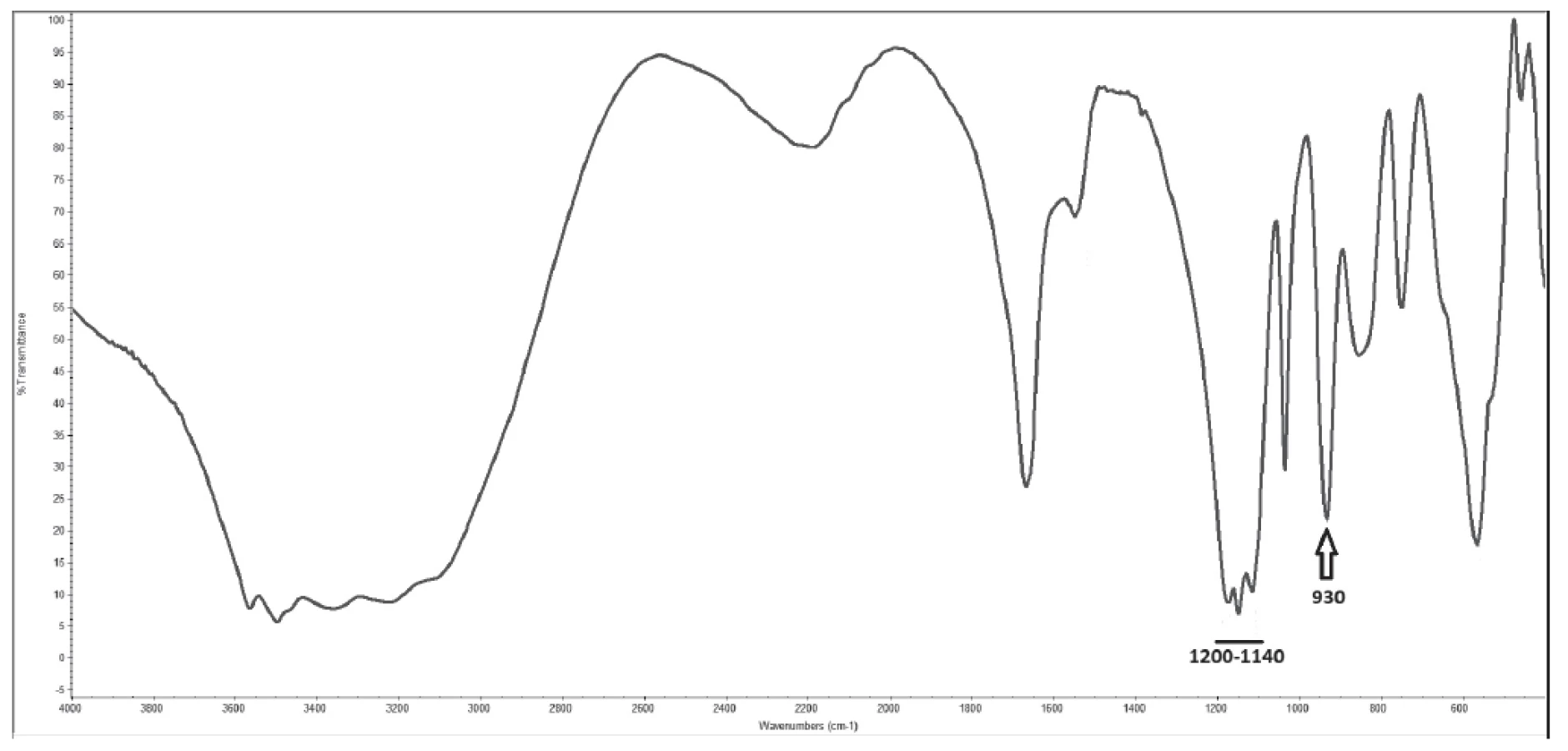
Velikost konkrementů byla 0,5-5x1-2x1-2 mm, barva bílá až nažloutlá, tvar nepravidelný s hladkým lesklým povrchem, tvrdost měkká až střední (obr. 1). Infračervená spektrometrie prokázala přítomnost pyrofosfátu vápenatého a síranu vápenatého v ekvimolárním množství (obr. 2) a polarizační mikroskopie anizotropní jehlicovité až soudečkovité krystaly s patrnými růstovými liniemi při okrajích (obr. 3). Specifické absorpční maximum pyrofosfátu se nachází v oblasti 930 cm-1 a sulfátu v oblasti 1200 – 1140 cm-1, obr. 2).


Diskuse
Perinatální forma má bez substituce chybějícího enzymu krajně nepříznivou prognózu, děti umírají pod obrazem respiračního selhání většinou v novorozeneckém období, maximálně do 1 roku věku [4, 15].
Našemu pacientovi je od druhého měsíce věku alkalická fosfatáza substituována. V biochemickém nálezu má hodnotu tohoto enzymu extrémně zvýšenou (na desetinásobek horní hranice referenčního rozmezí), což ukazuje na efektivitu terapie, takto vysoké hodnoty se považují za žádoucí. Léčba chlapci významně pomohla v kostních projevech nemoci, zlepšil se jeho růst (v pěti letech měří téměř 100 cm a váží 15 kg, což je považováno za dolní hranici fyziologických hodnot), došlo k postupné kostní mineralizaci, nedošlo k žádným frakturám. Zmírnily se též respirační obtíže, ventiluje spontánně přes tracheostomii, snížila se frekvence pneumonií.
Psychomotorický vývoj je však významně retardován, v pěti letech odpovídá maximálně přelomu I. a II. trimenomu, v pátém roce věku došlo navíc k progresi neurologického postižení. Na základě nejnovějších (dosud nepublikovaných) poznatků u podobně závažně postižených dětí ze zahraničí lze usuzovat, že tyto neurologické projevy není možné substituční enzymovou léčbou ovlivnit a lze předpokládat spíše další postupné zhoršování neurologického nálezu.
Substituovaná ALP nezabránila ani vzniku pyrofosfátových močových konkrementů, přestože jednou z hlavních funkcí ALP je konverze pyrofosfátu na fosfát. Další pozoruhodností v naší kazuistice je fakt, že právě pyrofosfát je obecně považován za inhibitor vzniku urolitiázy [2, 3].
Tyto neobvyklé pyrofosfátové konkrementy popsali ve svém článku též Frank A. et al., a to u čtyř koček a psa [20]. Pomocí spektrometrické analýzy prokázali přítomnost specifického absorpčního maxima charakteristického pro přítomnost pyrofosfátu v oblasti 930 cm-1. Autoři předpokládají, že sledovaná zvířata trpí metabolickou poruchou podobné hypofosfatázii u lidí. Tato porucha vedla k hypersaturaci moče pyrofosfátem, s následnou tvorbou do té doby nepopsaných močových konkrementů [20]. Stejné absorpční maximum svědčící pro přítomnost pyrofosfátu bylo opakovaně potvrzeno při analýzách konkrementu u pacienta prezentovaného v naší práci.
V období nálezu předchozího konkrementu tvořeného kyselinou močovou a urátem amonným dosahovalo pH moči maximálně hodnoty 6,5. V anamnestických laboratorních nálezech se nevyskytovala hyperurikemie, naopak spíše nižší hodnoty kyseliny močové. Terapeuticky bylo v té době doporučeno: alkalizovat moč směsí citronanu sodného a draselného (udržovat její pH mezi 6,5 a 6,9), podávat antiuratikum Alopurinol, a to i v případě normální urikemie, podávat dostatek tekutin. Od září 2018 (tj. v období před plánovanými litotrypsemi) bylo doporučeno profylaktické zajištění pacienta kotrimoxazolem.
V době nálezu pyrofosfátových konkrementů bylo pH moče 7,0. Alkaličtější moč byla pravděpodobně příčinou změny v biochemickém složení konkrementů. Pro alkalickou moč je typická tvorba fosfátových konkrementů, které však u našeho pacienta zjištěny nebyly. Vznik pyrofosfátového konkrementu bychom mohli hypoteticky vysvětlit nedostatečnou účinností rekombinantní ALP v ledvinách při konverzi pyrofosfátů na fosfáty. Pyrofosfáty jsou sice popisovány jako inhibitory krystalizace a agregace konkrementů, ale je možné, že přesycení alkalické moči pyrofosfátem indukuje naopak jejich tvorbu. Tomu nasvědčují i nálezy u zmíněných ALP deficientních zvířat s vysokou koncentrací pyrofosfátu v moči [20], kdy došlo k tvorbě pyrofosfátových konkrementů.
Přítomnost sulfátu v konkrementu byla velmi pravděpodobně způsobena podáváním sulfonamidů. Pacient užíval profylakticky Biseptol tbl 400 mg/80mg (kotrimoxazol). Jedná se o chemoterapeutikum složené ze sulfametoxazolu a trimetoprimu v poměru 5 : 1. Sulfametoxazol je N1-(5-metyl-3-isoxazolyl)sulfanilamid (C10H11N3O3S). Sulfonamidy mohou při snížení diurézy precipitovat v ledvinách a jsou považovány za významné litogenní faktory [21].
Pyrofosfáty a sulfáty komplexovaly s kalciovými ionty. V moči byla nalezena kalciurie na horní hranici referenčního rozmezí. Dalším patologickým nálezem u popisovaného pacienta byla proteinurie, která mohla být důsledkem právě urolitiázy.
Klinické zkušenosti s podáváním rekombinantní ALP u pacientů s hypofosfatázií jsou zatím omezené, nicméně studie z posledních let ukazují na jasné zlepšení stavu dětí s touto nemocí [4, 22]. Největší profit ze substituční terapie budou mít pravděpodobně děti s infantilní formou HPP, pro které je tato možnost život zachraňující [4, 23]. Otázkou zůstává dlouhodobý vývoj dětí s perinatální formou, řada z nich i přes zavedenou léčbu zůstává polymorbidní se spornou prognózou [4], jak je zřejmé také z naší kazuistiky.
Závěr
Předkládaná kazuistika popisuje klinický obraz a laboratorní nálezy u pětiletého chlapce s perinatální formou kongenitální hypofosfatázie a především poukazuje na nález neobvyklého močového konkrementu složeného z pyrofosfátu a sulfátu.
Střet zájmů: Autoři prohlašují, že nejsou ve střetu zájmů.
Do redakce došlo 18. 3. 2019
Adresa pro korespondenci:
Mgr. Milan Polák
Ústav lékařské chemie a klinické biochemie 2. LF UK a FN Motol
V Úvalu 84, 150 06 Praha 5
e-mail: milan.polak@fnmotol.cz
Zdroje
1. Jobs, K., Rakowska, M., Paturej, A. Urolithiasis in the pediatric population - current opinion on epidemiology, patophysiology, diagnostic evaluation and treatment. Dev Period Med., 2018, 22 (2), p. 201–208.
2. Sobotka, R., Hanuš, T. Příčiny a rizikové faktory vzniku urolitiázy. Urol. praxi, 2012, 13 (1), p. 11–15.
3. Moochhala, S. H., Sayer, J. A., Carr, G., Simmons, N. L. Renal calcium stones: insights from the control of bone mineralization Exp. Physiol., 2007, 93 (1), p. 43–49.
4. Šumník, Z., Souček, O., Lebl, J. Hypofosfatázie: Kdy na ni myslet a jak ji léčit. Pediatr. praxi, 2016, 17 (3), p. 146–149.
5. The Tissue Nonspecific Alkaline Phosphatase, Gene Mutations Database, SESEP Laboratory (Versailles Hospital - Unit of Constitutional Genetics) and the Unit of Cell and Genetics, University of Versailles-Saint Quentin en Yvelines, France [online]. 2019-02-10 [cit. 2019-01-15]. Accessible on WWW: <https://www.sesep.uvsq.fr/03_hypo_mutations.php>
6. Mornet, E. Hypophosphatasia. Orphanet J. Rare Dis., 2007, 2 (1), p.1–8.
7. Whyte, M. P. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev., 1994, 15 (4), p. 439–461.
8. Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon Med. Sch., 2010, 77 (1), p. 4 – 12.
9. Kodíček Milan Biochemické pojmy – výkladový slovník. Difosfát. Verze 1.0 Praha [cit. 2019-01-15]. Dostupný na WWW:<https://vydavatelstvi-old.vscht.cz/knihy/uid_es-002_v1/hesla/difosfat.html>. ISBN 80-7080-551-X.
10. Rachow, J. W., Ryan, L. M. Inorganic pyrophosphate metabolism in arthritis. Rheum. Dis. Clin. North. Am., 1988, 14 (2), p. 289–302.
11. Russell, R. G. Metabolism of inorganic pyrophosphate (PPi). Arthritis Rheum., 1976, 19 (Suppl. 3), p. 465–478.
12. Jung, A., Russel, R. G., Bisaz, S., Morgan, D. B., Fleisch, H. Fate of intravenously injected pyrophosphate-32 P in dogs. Am. J. Physiol., 1970, 218 (6), p. 1757–1764.
13. Nouwen, E. J., De Broe, M. E. Human intestinal versus tissue-nonspecific alkaline phosphatase as complementary urinary markers for the proximal tubule. Kidney Int., 1994, 47, p. 43 – 51.
14. Hypophosphatasia Signs and Symptoms. [online]. 2014-09-10 [cit. 2019-01-15]. Dostupný na WWW: <https//hypophosphatasia.com>
15. Chaloupek, K., Bartoníková, A., Souček, O., Šumník, Z., Vlk, R., Černý, M. Perinatální forma hypofosfatázie – kazuistika (abstrakt). Neonatologické listy, 2014, 20 (2), p. 39.
16. Šumník, Z., Černý, M., Bačkai, T., Padidela, R., Souček, O. Kolik stojí letenka do Manchesteru? (abstrakt). Osteol. Bull., 2015, 20 (2), p. 90.
17. Metz, M. P. Determining urinary calcium/creatinine cut-offs for the paediatric population using published data. Ann. Clin. Biochem., 2006, 43, p. 398–401.
18. Matos, V., van Melle, G., Boulat, O., Markert, M., Bachmann, C., Guignard, J. P. Urinary phosphate/creatinine, calcium/creatinine, and magnesium/creatinine ratios in a healthy pediatric population. J. Pediatr., 1997, 131 (2), p. 252–257.
19. Simplified Infrared Correlation Chart University of Wisconsin, Department of chemistry [online]. [cit. 2019-01-15]. Accessible on WWW: <https://www.chem.wisc.edu/deptfiles/OrgLab/handouts/Simplified%20IR%20Correlation%20Chart.pdf>
20. Frank, A., Norrestam, R., Sjödin, A. A new urolith in four cats and a dog: composition and crystal structure. J. Biol. Inorg. Chem., 2001, 7 (4–5), p. 437–444.
21. FDA – BactrinTM [online]. [cit. 2019-01-15]. Accessible on WWW: <https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/017377s068s073lbl.pdf
22. Whyte, M. P., Greenberg, C. R., Salman, N. J. et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med., 2012, 366 (10), p. 904–913.
23. Whyte, M. P., Rockman-Greenberg, C., Ozono, K. et al. Asfotase Alfa treatment improves survival for perinatal and infantile hypophosphatasia. J. Clin. Endocrinol. Metab., 2016, 101 (1), p. 334–342.
Štítky
Biochémia Nukleárna medicína Nutričný terapeutČlánok vyšiel v časopise
Klinická biochemie a metabolismus

2019 Číslo 2
Najčítanejšie v tomto čísle
- Porovnání výsledků vysoce senzitivních stanovení troponinů I (Abbott, Beckman Coulter, Siemens) a troponinu T (Roche)
- POCT systém detekce hladin fekálního kalprotektinu při telemonitoringu pacientů s idiopatickými střevními záněty
- Změny močových a sérových markerů poškození ledvin u dospělých pacientů po podání kontrastní látky při angiografii
- Elongázy mastných kyselin a jejich účast v patogeneze chorobných stavů