
Myokarditida a kardiomyopatie z pohledu kardiologa
Clinical perspective on the myocarditis and cardiomyopathies
Myocardial diseases are often encountered in cardiology and pose a significant diagnostic challenge. Myocarditis is an acute inflammatory disease of the heart muscle. Pathophysiology of myocarditis is a complex interplay of genetic background, innate immunity, viral or bacterial agents and formation of autoreactive antibodies and lymphocytes that maintain the inflammation after the infection was eliminated. Differentiation of myocardial infarction or heart failure of different etiology is crucial in the acute stage. Cardiac magnetic resonance imaging (MRI) enables with sufficient sensitivity and specificity diagnosis of myocardial inflammation and scar. Endomyocardial biopsy (EMB) with histology and immunohistochemistry is a gold standard for detection of myocarditis. EMB is indicated in selected patients with life-threatening symptoms where EMB may have therapeutic consequences. Giant cell myocarditis and eosinophilic myocarditis are specific examples of such a condition. Polymerase chain reaction (PCR) of the myocardial sample is used to detect viral genome. Serum antibodies or PCR from blood are not helpful in determining the etiology of myocarditis. Viral presence in myocardium is found in patients who do not have histological evidence of myocarditis which makes the association of positive PCR and etiology of myocarditis obscure. Cardiomyopathies (CMP) are characterized by structural and functional cardiac abnormalities that cannot be explained by coronary artery disease or abnormal loading conditions (valvular disease, arterial hypertension, congenital heart disease). CMP are classified based on the prevailing morphology regardless of primary (genetic, idiopathic) or secondary (systemic disease) etiology. European Society of Cardiology defines five types of CMP: hypertrophic, dilated, restrictive, arrhythmogenic and unclassified. CMP diagnosis is based on the imaging with echocardiography, coronary angiography, invasive hemodynamics and cardiac MRI. EMB is rarely indicated in dilated or restrictive CMP. Genetic testing is used to determine pathogenic mutations in phenotype positive patients and in familiar screening. Genetically determined CMP are mostly monogenic and autosomal dominant. Incomplete penetrance and variable expressivity cause variable or even negative phenotypes in genotype positive individuals. Genetic screening of a large number of genes and non-coding DNA results in findings of many variants of uncertain significance which make the interpretation of the genetic testing difficult.
Keywords:
myocarditis – clinical diagnosis of myocarditis – endomyocardial biopsy – cardiomyopathy – classification of cardiomyopathies – genetics of cardiomyopathies
Autoři:
Cyril Štěchovský 1; Theodor Adla 2
![]() ; MUDr. Jiří Bonaventura, Ph.D. 1
; MUDr. Jiří Bonaventura, Ph.D. 1
Působiště autorů:
Kardiologická klinika 2. LF UK a Fakultní nemocnice v Motole, Praha
1; Klinika zobrazovacích metod 2. LF UK a Fakultní nemocnice v Motole, Praha
2
Vyšlo v časopise:
Čes.-slov. Patol., 55, 2019, No. 4, p. 209-217
Kategorie:
Přehledový článek
Souhrn
Onemocnění myokardu představují častou, a zároveň obtížnou, diferenciální diagnostiku v klinické kardiologii. Myokarditida je akutní zánětlivé onemocnění myokardu. Ke vzniku myokarditidy a jejímu průběhu přispívá genetická predispozice, schopnost eliminace virového nebo mikrobiálního agens, tvorba autoprotilátek proti srdečním strukturám a tvorba autoagresivních lymfocytů, které udržují zánět i po odeznění akutní infekce. Základem diferenciální diagnostiky je odlišení od infarktu myokardu nebo srdečního selhávání jiné etiologie. Magnetická rezonance srdce umožňuje s dobrou senzitivitou i specificitou zobrazení zánětu v myokardu, nikoliv však jeho etiologie. V indikovaných případech provádíme endomyokardiální biopsii (EMB), která je zlatým standardem diagnostiky myokarditidy. Histologické a imunohistochemické vyšetření odliší specifické typy myokarditidy, jako jsou obrovskobuněčná a eosinofilní myokarditida, které mají často fatální průběh, a pro které existuje specifická terapie. Molekulárně genetické vyšetření pomocí polymerázové řetězové reakce (PCR) z EMB se využívá k detekci virového nebo bakteriálního genomu v myokardu. Sérologie ani PCR z krve nejsou dostatečně specifické k určení etiologie virové myokarditidy. V případě některých virů není jasná asociace mezi přítomností virového genomu v myokardu a etiologií myokarditidy, virový genom je nalézán i u pacientů, kteří zjevně myokarditidou netrpí. Efekt imunosupresivní léčby lymfocytární myokarditidy je nadále předmětem klinických studií. Kardiomyopatie (KMP) jsou skupinou onemocnění se strukturálním a funkčním postižením myokardu, které není způsobeno ischemickou chorobou srdeční nebo abnormálními podmínkami preloadu a afterloadu (chlopenní vady, arteriální hypertenze, vrozené srdeční vady). KMP klasifikujeme na základě jejich morfologie bez ohledu na etiologii. Klasifikace Evropské kardiologické společnosti rozlišuje pět skupin KMP: hypertrofickou, dilatační, restrikční, arytmogenní a neklasifikovanou KMP. K diagnostice využíváme echokardiografii, koronarografii, katetrizační hemodynamické vyšetření a magnetickou rezonanci. EMB je výjimečně indikována u dilatační a restrikční KMP. Genetické vyšetření slouží k identifikaci patogenních mutací u jedinců s vyjádřeným fenotypem a k screeningu příbuzných. Většina geneticky podmíněných KMP jsou monogenní onemocnění s autozomálně dominantním přenosem. Nekompletní penetrance a variabilní expresivita způsobují rozdílný fenotypový projev nebo negativní fenotyp u geneticky pozitivních jedinců. Genetický screening velkého počtu genů má za následek časté nálezy variant nejasného významu, které činí interpretaci genetického vyšetření velmi komplikovanou.
Klíčová slova:
myokarditida – klinická diagnostika myokarditidy – endomyokardiální biopsie – kardiomyopatie – klasifikace kardiomyopatií – genetika kardiomyopatií
S onemocněním myokardu se v klinické kardiologii setkáváme často, myokarditida patří mezi základní diferenciální diagnostiku bolestí na hrudi a akutního srdečního selhání, zároveň však představuje obtížný diagnostický úkol, hypertrofická kardiomyopatie je jedním z nejčastějších genetických onemocnění, dilatační kardiomyopatie je častou příčinou srdečního selhávání a nejčastější indikací k srdeční transplantaci. Následující text shrnuje základní poznatky o patofyziologii, etiologii a diagnostice myokarditidy a kardiomyopatií. Léčebné možnosti jsou zmíněny jen okrajově. Text je koncipován z pohledu klinického kardiologa – od symptomů, přes vyšetřovací metody používané a dostupné v kardiocentrech, ke stanovení diagnózy. Jsou zmíněny jen zásadní klasifikační systémy a diagnostická kritéria, které umožňují lékařům různých specializací i národností hovořit stejnou řečí. Nejedná se o komplexní shrnutí patofyziologie ani etiologie onemocnění myokardu ani o doporučení odborných společností. Je kladen důraz na využití vyšetřovacích metod, kterými disponuje moderní patologie – od histologie až po molekulární biologii. V textu jsou dále shrnuty současné poznatky o genetice kardiomyopatií. Diagnostika onemocnění myokardu vyžaduje multioborovou spolupráci kardiologa, radiologa, patologa, klinického genetika a dalších odborností. Z tohoto důvodu je v tomto čísle časopisu publikován článek zabývající se problematikou myokarditidy a kardiomyopatií z pohledu patologa, kde čtenář nalezne podrobné informace týkající se histologického a imunohistochemického vyšetření u onemocnění myokardu a dalších molekulárních mechanismů, které nejsou uvedeny v tomto textu.
MYOKARDITIDA
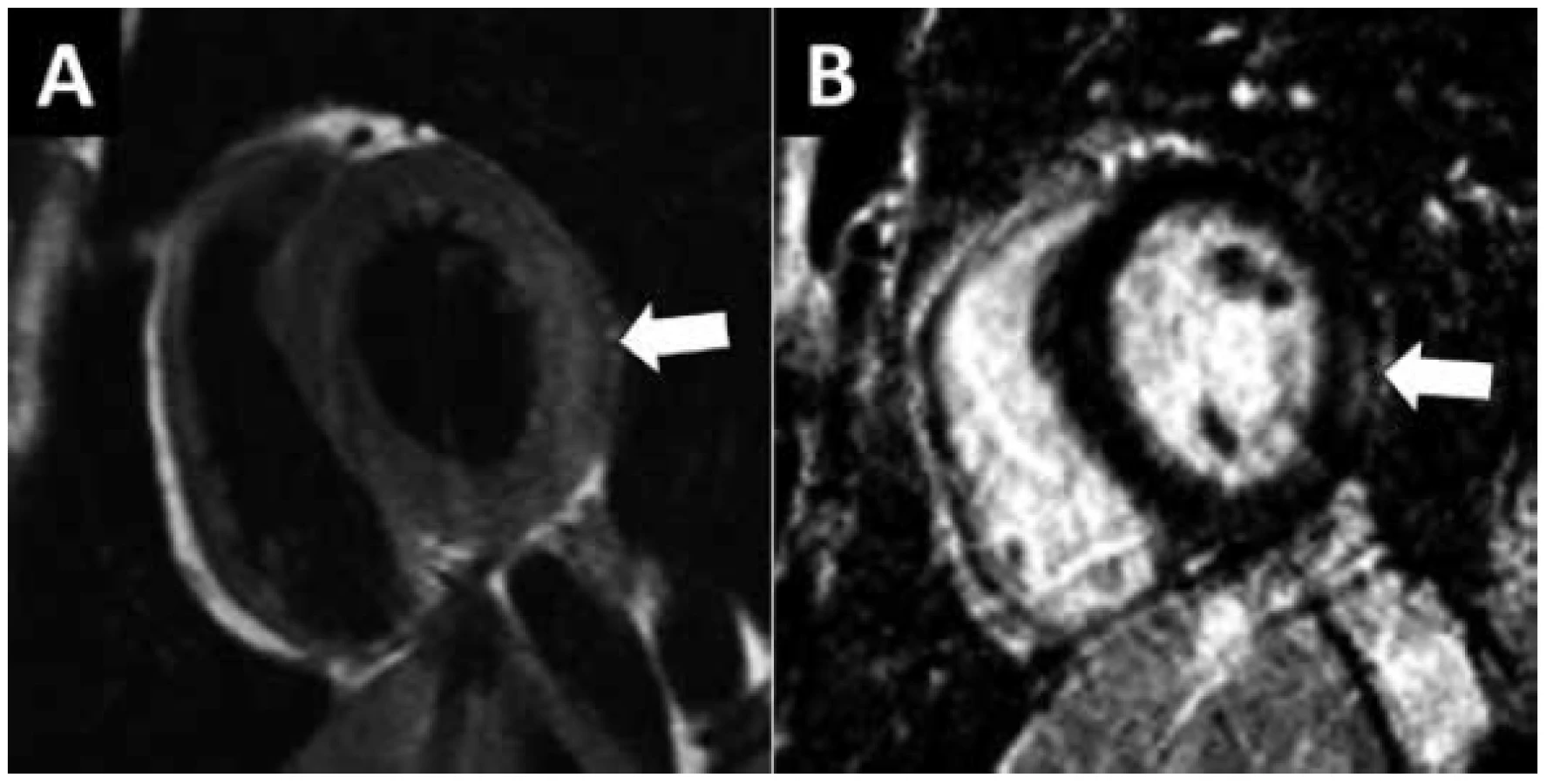
V nejširším smyslu je myokarditida každé zánětlivé onemocnění myokardu (1). Klinicky definujeme myokarditidu jako akutní onemocnění myokardu způsobené zánětlivou odpovědí na infekční (virové, bakteriální, parazitární), toxické (včetně lékového) nebo autoimunní agens (2). Symptomy myokarditidy nejsou specifické a zahrnují febrílie, infekci horních dýchacích cest („chřipkové příznaky“), bolesti na hrudi, srdeční selhávání, arytmie a náhlou smrt. Stejně tak laboratorní a zobrazovací vyšetření dostupná u lůžka pacienta nejsou dostatečně specifická pro diagnostiku myokarditidy. Pravidlem je různá míra elevace srdečních troponinů nad 99. percentil normálních hodnot, podle hladiny troponinu (deseti až tisícinásobná elevace) však nelze odlišit myokarditidu od jiných příčin poškození myokardu. Elektrokardiogram (EKG) může být normální nebo jsou přítomny změny repolarizace imitující ischémii, poruchy převodu nebo supraventrikulární a komorové arytmie. Echokardiograficky je, podle závažnosti postižení, normální struktura i funkce srdce nebo je přítomna systolická a diastolická dysfunkce obou komor, která se může, i v průběhu dnů, dramaticky zhoršovat. Přítomnost perikardiálního výpotku svědčí pro současné zánětlivé postižení perikardu – perimyokarditidu. Tíže systolické dysfunkce levé komory (LK) koreluje se závažností postižení a s prognózou pacienta (3). Na prvním místě je nutné odlišit myokarditidu od jiných příčin postižení myokardu, které se mohou prezentovat stejnými klinickými příznaky, elevací srdečních troponinů, EKG změnami a echokardiografickým obrazem dysfunkce LK. Prakticky ve všech případech provádíme koronarografii nebo CT-koronarografii k vyloučení infarktu myokardu. Dle klinického stavu dále obvykle provedeme vyšetření srdce magnetickou rezonancí (MR). Při splnění tzv. Lake Louise kritérií (4) je MR srdce senzitivní i specifickou metodou k průkazu akutní myokarditidy (Obr. 1) a zároveň odlišení od infarktu myokardu se spontánní rekanalizací koronární trombózy. Na základě výše uvedených klinických symptomů, laboratorních a zobrazovacích vyšetření stanovujeme klinickou diagnózu myokarditidy (Tab.1). K definitivnímu potvrzení myokarditidy a její etiologie je zapotřebí histologické vyšetření myokardu doplněné o imunohistochemické a molekulárně genetické vyšetření. Tomuto tématu je věnována samostatná publikace. Myokarditida diagnostikovaná per exclusionem, bez provedení histologického vyšetření nebo bez známek zánětu myokardu při vyšetření MR, by měla zůstat pracovní diagnózou.

Endomyokardiální biopsie (EMB) z pravé komory se provádí cestou v.femoralis nebo v. jugularis interna pod echokardiografickou a/nebo skiaskopickou kontrolou, biopsie z LK se provádí velmi zřídka pro riziko systémové embolizace. EMB je zatížena 1-2% rizikem vzniku srdeční tamponády, což je život ohrožující komplikace. Při klinickém podezření na myokarditidu je EMB indikována jen v případech, kdy histologická a etiologická specifikace myokarditidy může zásadním způsobem ovlivnit terapii. Při indikaci k EMB se řídíme doporučeními Evropské kardiologické společnosti (5). K EMB jsou indikováni pacienti s akutním srdečním selháváním a/nebo život ohrožujícími arytmiemi, při vyloučení jiných příčin (ischemická choroba srdeční, chlopenní vady), s těžkou a/nebo rychle progredující systolickou dysfunkcí LK a s krátkou anamnézou srdečního selhávání (do 3 měsíců). EMB může být v individuálních případech zvážena i u pacientů s nově zjištěnou dilatační kardiomyopatií (DKMP), kteří nesplňují výše uvedená kritéria, a u kterých není pravděpodobná jiná příčina vzniku DKMP (abúzus alkoholu, toxicita chemoterapie). EMB odliší ostatní formy DKMP od zánětlivé DKMP charakterizované chronickým zánětlivým infiltrátem, který nalézáme u 9 % dospělých pacientů s DKMP a u 46 % dětí s DKMP (2). Naopak u pacientů s klinickým podezřením na myokarditidu a s benigním průběhem (dobrá funkce LK nebo spontánní zlepšení, rychlé vymizení symptomů a pokles troponinu) je klinický přínos EMB velmi malý. U pacientů s chronickým srdečním selháváním s dlouhou anamnézou nemoci není EMB přínosná. Z klinického pohledu je zásadní odlišit lymfocytární myokarditidu (virovou nebo idiopatickou) od obrovskobuněčné myokarditidy a eosinofilní myokarditidy. Tyto dvě entity jsou spojeny s velmi závažným průběhem a mají specifickou imunosupresivní léčbu. Granulomatózní zánět myokardu při sarkoidóze, který je klinicky označován jako srdeční sarkoidóza, je některými autory (2) považován také za myokarditidu. Zánětlivý infiltrát při srdeční sarkoidóze bývá fokální, nejčastěji v interventrikulárním septu, a může vést k falešně negativní EMB. Srdeční sarkoidóza je diagnostikována pomocí zobrazení ložisek na MR srdce a jejich aktivity na pozitronové emisní tomografii (PET). Klinický přínos detekce virového genomu z EMB zatím není jednoznačně potvrzen, nicméně molekulárně genetické vyšetření z EMB bývá obvykle prováděno. Na dvou studiích s malým počtem pacientů se prokázalo mírné zlepšení funkce LK u akutní myokarditidy (6) respektive zánětlivé DKMP (7) při absenci virového genomu v myokardu a imunosupresivní léčbě.
Etiologie myokarditidy je velice pestrá (2) (Tab. 2), vzhledem k nutnosti provedení EMB a nákladných molekulárně genetických vyšetření však zůstává v individuálních případech s benigním průběhem obvykle neobjasněna. Taková myokarditida bývá označována jako idiopatická, což je zřejmě vhodnější, než označení virová, pokud detekce virového genomu z myokardu nebyla provedena. Sérologické ani molekulárně genetické vyšetření z krve není dostačující k průkazu virové myokarditidy, neboť séropozitivita a přítomnost DNA nebo RNA virů asociovaných s myokarditidou je častá i u zdravých osob (8). Také molekulárně genetický průkaz virové DNA nebo RNA v myokardu bez histologického průkazu myokarditidy nemá jasný klinický význam a nelze ho považovat za průkaz etiologie dysfunkce LK, neboť výskyt virového genomu v myokardu u pacientů podstupujících srdeční operaci (koronární bypass, operaci chlopně atd.) je také častý (9). Tyto nálezy zpochybňují příčinnou souvislost například mezi parvovirem B19 a myokarditidou (10). Klinická diagnóza myokarditidy ve spojení s průkazem borrelií (Lymeská karditida), leptospir (Weilova nemoc), Trypanosoma cruzi (Chagasova nemoc) a jiných mikroorganismů asociovaných s myokarditidou je dostatečná k určení etiologie a zahájení specifické antimikrobiální léčby. Řada systémových autoimunních onemocnění se může projevovat také postižením myokardu (Tab. 2), výjimečně může být myokarditida prvním nebo jediným projevem těchto onemocnění. Léčba cílená na základní onemocnění vede v těchto případech také k potlačení myokarditidy. Ke vzniku myokarditidy a jejímu průběhu přispívá genetická predispozice, schopnost eliminace virového nebo mikrobiálního agens, tvorba autoprotilátek proti srdečním strukturám a tvorba autoagresivních lymfocytů, které udržují zánět i po odeznění akutní infekce (2).

Průběh myokarditidy je variabilní, lze pozorovat několik „klinických fenotypů“, které se mohou kombinovat nebo přecházet z jednoho do druhého u téhož pacienta. Častá je prezentace symptomy akutního infarktu myokardu s EKG změnami a pozitivitou troponinu, kdy dochází k rychlému vymizení symptomů, funkce LK je zachována nebo dojde časně k její normalizaci.U těchto pacientů ordinujeme dočasný klidový režim a kontrolní echokardiografii, farmakoterapie není indikována a prognóza je dobrá. Dalším fenotypovým projevem myokarditidy je akutní srdeční selhávání se sníženou ejekční frakcí (EF) LK, které může být doprovázeno závažnými arytmiemi. U těchto pacientů obvykle provádíme EMB a ordinujeme dlouhodobou farmakoterapii srdečního selhávání a kardiologickou dispenzarizaci. Prognóza je závislá na vývoji EFLK, může dojít k normalizaci nebo rozvoji zánětlivé DKMP. Efekt imunosupresivní léčby u těchto pacientů je nadále předmětem klinických studií. Zatím největší provedená randomizovaná studie zkoumající efekt kombinované imunosupresivní léčby u akutní biopticky prokázané myokarditidy však dopadla negativně, nedošlo ke zlepšení funkce LK a nebyla ovlivněna ani pětiletá mortalita (11). Je možné, že pacienti, u kterých došlo k eliminaci virového genomu, mají lepší prognózu (6,7,12,13). Myokarditida může probíhat také fulminantně s rozvojem kardiogenního šoku, v těchto případech používáme stejné léčebné metody, jako při akutním srdečním selhání jiné etiologie, včetně mechanické podpory oběhu a srdeční transplantace. Dalším projevem myokarditidy je náhlá smrt, myokarditida je nalézána u 2-12% mladých osob zemřelých náhlou smrtí (14,15).
KLASIFIKACE KARDIOMYOPATIÍ
V klinické medicíně pod pojmem kardiomyopatie (KMP) rozumíme strukturální a funkční postižení myokardu, které není způsobeno ischemickou chorobou srdeční (ICHS) nebo abnormálními podmínkami preloadu a afterloadu, což v praxi znamená chlopenními vadami nebo arteriální hypertenzí. Klasifikace kardiomyopatií prošla dlouhým vývojem. Původní dělení na primární onemocnění myokardu neznámé příčiny, které byly klasifikovány dle morfologie na hypertrofickou, dilatační a restrikční KMP, a sekundární KMP při onemocněních se systémovým postižením (např. střádavá onemocnění, svalové dystrofie atd.) nebo jako následky působení toxinů, léků, radioterapie, se s rozvojem poznatků o genetice a molekulárních mechanismech ukázalo být nepřesné (16). Především byla identifikována řada genů a velké množství mutací, které jsou zodpovědné za rozvoj hypertrofické, dilatační a arytmogenní KMP (17-19). Jistá nejednoznačnost v názvosloví kardiomyopatií a různé klasifikační systémy napříč Atlantikem však nadále trvají. Situaci dále komplikuje v anglické literatuře běžné používání termínu ischemická kardiomyopatie, označující systolickou dysfunkci LK na podkladě ICHS, a zařazení geneticky podmíněných poruch transmembránových kanálů (kanálopatií) mezi KMP v severoamerické klasifikaci. Klasifikace Evropské kardiologické společnosti (ESC) z roku 2008 (17) se drží především morfologického dělení KMP a rozlišuje pět hlavních skupin KMP: hypertrofickou, dilatační, restrikční, arytmogenní kardiomyopatii pravé komory (ARVC) a neklasifikovanou KMP. Každá KMP je dále dělena na familiární (dědičnou) a získanou formu (Tab. 3). Většina geneticky podmíněných KMP jsou monogenní onemocnění, vyskytující se sporadicky (de novo mutace) nebo familiárně v pravém smyslu, obvykle s autozomálně dominantním přenosem. Ani přítomnost genetické mutace však nezaručuje fenotypový projev a nekoreluje s jeho závažností např. u nositelů téže mutace. Tento jev se nazývá variabilní expresivita a neúplná penetrance a není do detailů objasněn. Při současné úrovni poznání, zůstane část případů, i po důkladném genetickém vyšetření, nezařazena. Výhodou evropského klasifikačního systému je relativní jednoduchost zařazení onemocnění dle dominantního fenotypu. K diagnostice kardiomyopatie je obvykle dostačující provedení echokardiografického vyšetření, které určí dominantní fenotyp a zároveň vyloučí významnou primární chlopenní vadu nebo vrozenou zkratovou vadu. V případě dilatační KMP je nezbytně nutné vyloučení významné koronární nemoci, v případě ARVC je diagnostický algoritmus složitější a zahrnuje poruchy depolarizace a repolarizace a vyšetření srdce magnetickou rezonancí. Nevýhodou fenotypové klasifikace KMP je skutečnost, že podobný fenotypový projev má různou etiologii (Tab. 4), prognózu a v některých případech i specifickou terapii. Například k srdečnímu selhání s obrazem dilatační KMP může vést familiární dilatační KMP, chronická myokarditida s rozvojem zánětlivé dilatační KMP, abusus alkoholu nebo řada dalších vzácnějších příčin. Dále je známo, že rozdílné mutace v témže genu mohou vést k fenotypově rozdílným projevům.


Dilatační kardiomyopatie
Dilatační KMP je charakterizována systolickou dysfunkcí dilatované levé komory při nepřítomnosti chlopenní vady, těžké arteriální hypertenze a koronární nemoci, která by mohla tuto dysfunkci způsobit. Dilatace a dysfunkce pravé komory a dilatace obou síní bývá u symptomatických pacientů také přítomna, ale není nezbytná pro diagnózu. Přesná prevalence není známa a je geograficky variabilní, v západoevropské populaci je odhadována na 1 : 2000 osob (17). Etiologie DKMP je různorodá (Tab. 4) a je také geograficky variabilní (např. Chagasova choroba v Jižní Americe nebo nutriční deficit selenu v některých oblastech severovýchodní Číny). V souborech pacientů z Evropy a Severní Ameriky se zastoupení pacientů s familiární formou pohybuje okolo 25 % (17). Autozomálně dominantní formy onemocnění jsou způsobeny mutacemi v genech pro cytoskelet, sarkomerické proteiny, proteiny Z-disku a desmozomy. X-vázané formy jsou asociovány s muskulární dystrofií. Získané formy DKMP jsou v naší populaci nejčastěji spojovány s toxicitou protinádorové terapie (antracykliny, cyklofosfamid, tyrozin-kinázové inhibitory a monoklonální protilátka trastuzumab), dlouhodobým abúzem alkoholu a následky myokarditidy. Zánětlivou DKMP nalézáme u 9 % dospělých pacientů s DKMP a u 46 % dětí s DKMP (17), u kterých byla po stanovení diagnózy DKMP provedena EMB. U některých pacientů nacházíme nedilatovanou hypokinetickou LK, může se jednat o počáteční stádia DKMP (např. při vyšetření asymptomatických příbuzných pacientů s DKMP) nebo o projev onemocnění při mutaci genu pro lamin A/C (20). V těchto případech je označení DKMP zavádějící a pracovní skupinou ESC bylo navrženo označení hypokinetická nedilatovaná KMP (21). Peripartální KMP je vzácnou formou DKMP vyskytující se během posledních 6 týdnů gravidity a až 5 měsíců postpartálně (22). Často dochází k normalizaci funkce LK, ale riziko recidivy při opakované graviditě je vysoké. Ke vzniku tachykardií indukované KMP může dojít již po několika týdnech trvání arytmie (nejčastěji fibrilace nebo flutteru síní) s rychlou komorovou frekvencí. Diagnóza tachykardií indukované KMP může být potvrzena až poté, co dojde k normalizaci funkce LK po úspěšné kontrole tepové frekvence nebo ukončení arytmie. Je zřejmé, že všechny výše zmíněné příčiny získané DKMP vyžadují ke svému projevu genetický podklad, neboť fenotyp DKMP vyvine jen menší část osob vystavených kardiotoxické chemoterapii, alkoholu nebo virové myokarditidě.
Klinickými projevy DKMP jsou srdeční selhávání, arytmie nebo náhlá smrt. Diagnóza je stanovena na podkladě echokardiografie, přičemž nelze rozlišit mezi DKMP a remodelovanou, dilatovanou a dysfunkční LK po proběhlém infarktu myokardu přední stěny, v obou případech může být přítomen apikální trombus a sekundární mitrální regurgitace. EKG při DKMP bývá téměř vždy abnormální, ale změny nejsou specifické, častá je blokáda levého raménka Tawarova. K odlišení od ICHS je nutné provést koronarografii. V některých případech provádíme i MR srdce (Obr. 2), kde může být přítomno pozdní sycení gadoliniem jako známka fibrózy myokardu, ale jeho rozložení je jiné, než v případě jizvy po subendokardiálním nebo transmurálním infarktu myokardu. Léčba se skládá z farmakoterapie srdečního selhávání, přístrojové léčby (resynchronizační léčba, implantabilní defibrilátory, mechanické srdeční podpory) a srdeční transplantace. Farmakoterapie nemá být ukončena, ani pokud dojde ke zlepšení či normalizaci funkce LK (23). DKMP je nejčastější příčinou srdeční transplantace v České republice.

Restrikční kardiomyopatie
Restrikční KMP (RKMP) není na rozdíl od DKMP a HCM definována typickým morfologickým obrazem ale fyziologií plnění LK. Restrikční plnění znamená, že dochází ke strmému nárůstu tlaku v LK v časné diastole a poté následuje fáze plató, kdy již nedochází ke zvyšování objemu LK. Restrikční hemodynamika není přítomna jen u RKMP, ale nalézáme ji i prakticky vždy při pokročilém srdečním selhávání jakékoliv etiologie. RKMP je tedy onemocnění, při kterém není zvětšený enddiastolický ani endsystolický objem LK a není přítomna výrazná hypertrofie LK (17). Kontraktilita LK je prakticky vždy snížená. Vzhledem k vysokému plnícímu tlaku LK a často i PK je přítomna výrazná dilatace obou síní a s tím související častý výskyt fibrilace síní. Prevalence RKMP není známa ale je nižší než hypertrofické a DKMP. Nejčastější příčinou RKMP je ukládání amyloidu do myokardu. Podle typu amyloidu rozeznáváme AL amyloidózu (lehké řetězce imunoglobulinů), která má obvykle ještě renální postižení, avšak postižení myokardu bývá klinicky dominantní. Při transtyretinové amyloidóze dochází k ukládání patologicky uspořádaných molekul transtyretinu (TTR). Existuje forma familiární (mutace v genu pro TTR), kdy je přítomna neuropatie a hepatopatie a forma senilní (wild-type TTR), kdy je kardiální postižení dominantní. Při srdeční amyloidóze je typické ztluštění atrioventrikulárních chlopní, síňového septa a malý až středně velký perikardiální výpotek. Vzácně se jako RKMP mohou manifestovat střádavá onemocnění jinak vedoucí k fenotypu HCM. RKMP může vzniknout jako následek endomyokardiální fibrózy při hypereosinofilním syndromu. RKMP je také jednou z manifestací primární i sekundární hemochromatózy. Není vzácným nálezem u pacientů léčených pro pokročilý myelodysplastický syndrom nebo chronickou leukémii se závislostí na transfúzní léčbě.
Klinickým projevem RKMP je bilaterální srdeční selhávání a fibrilace síní. Diagnóza je stanovena na podkladě echokardiografie obvykle doplněné o katetrizační hemodynamické vyšetření. MR srdce odliší přetížení železem při hemochromatóze, obraz srdeční amyloidózy není zcela specifický (Obr. 3). AL amyloidóza je diagnostikována hematologicky. Transtyretinovou amyloidózu lze detekovat pomocí pyrofosfátové scintigrafie. K definitivnímu potvrzení amyloidózy může být provedena EMB. Prognóza srdeční amyloidózy je závažná a léčebné možnosti jsou limitované.

Arytmogenní kardiomyopatie pravé komory
Diagnóza arytmogenní kardiomyopatie pravé komory (ARVC) je stanovena na základě komplexních kritérií (24) hodnotících anamnézu, arytmie, poruchy depolarizace a repolarizace a morfologii a funkci PK (Obr. 4). Při ARVC dochází k progresivní náhradě myokardu PK tukovou a fibrózní tkání s rozvojem dilatace a dysfunkce PK a vznikem aneurysmatu PK. Obdobné změny byly u ARVC popsány i v LK. Přes tyto morfologické změny PK nepoužíváme EMB k diagnostice onemocnění, riziko falešně pozitivních i negativních biopsií a vlastní riziko EMB z dilatované a ztenčené PK je vysoké. Prevalence onemocnění se odhaduje na 1 : 5000. Většina forem ARVD jsou autozomálně dominantně dědičné mutace genů pro desmosomální proteiny.

Klinický průběh onemocnění má tří fáze, v prvním stádiu nejsou přítomny typické morfologické změny PK ani EKG změny a jediným projevem mohou být komorové arytmie a náhlá smrt, ve druhém stádiu je již přítomna remodelace PK s typickými EKG změnami a jsou časté komorové arytmie, ve třetím stádiu dochází k selhání PK. Vzhledem k tomuto průběhu je ARVD nalézána dle některých autorů jako relativně častá příčina náhlé smrti mladých osob (25). Léčba spočívá v identifikaci rizika náhlé smrti a implantaci defibrilátoru, komorové arytmie lze někdy ovlivnit katetrizační ablací, při refrakterních arytmiích nebo srdečním selhávání je jedinou možností mechanická oběhová podpora nebo transplantace srdce.
Hypertrofická kardiomyopatie
Hypertrofická kardiomyopatie (HCM) je onemocnění charakterizované jinak nevysvětlitelnou hypertrofií srdeční svaloviny (18,26). Ve většině případů je považována za hereditární onemocnění s nejčastěji autosomálně dominantní dědičností (27). S frekvencí výskytu 1 : 500 až 1 : 200 se řadí mezi nejčastější dědičná srdeční onemocnění a jedná se pravděpodobně o nejčastější příčinu náhlé srdeční smrti u mladých sportovců (28,29).
Základním klinickým diagnostickým kritériem je přítomnost tloušťky srdeční svaloviny ≥ 15 mm zjištěné zobrazovacím vyšetřením (26,30). V klinické praxi používáme nejčastěji echokardiografické vyšetření, které je levné a dostupné. U vybraných pacientů a v rámci rizikové stratifikace je indikována MR srdce. U prvostupňových příbuzných pacienta s již diagnostikovanou HCM jsou kritéria přísnější; k diagnóze stačí, po vyloučení hemodynamických příčin, tloušťka jakéhokoli segmentu stěny LK ≥ 13 mm (26,30). Pro diagnózu HCM je dostatečná hypertrofie v jakémkoliv segmentu LK, asymetrická septální hypertrofie je častější než koncentrická hypertrofie LK. Nejčastější je postižení bazálních a středních anteroseptálních segmentů. Rozsah hypertrofie je velmi variabilní a její tíže je spojena s rizikem náhlé smrti (18). V klinické praxi je obtížné odlišit pomocí neinvazivních zobrazovacích metod pravou hypertrofii kardiomyocytů od zesílení stěn podmíněného intersticiální fibrózou, infiltrací nebo akumulací metabolických substrátů. Proto současná definice ESC nepovažuje přítomnost akumulace patologického substrátu v myokardu za vylučující kritérium a definuje HCM přítomností zesílené stěny nebo celkové hmotnosti jedné či obou srdečních komor při vyloučení hemodynamické abnormality, která by mohla danou hypertrofii způsobit (17,26). Mezi nejčastěji takto vylučované diagnózy patří arteriální hypertenze, aortální stenóza, koarktace aorty a tzv. atletické srdce. V diagnostice pacientů s HCM má stále větší uplatnění MR srdce (Obr. 5). Dokáže přesně zhodnotit přítomnost, rozsah a tíži hypertrofie myokardu. MR má též význam v diagnostice méně obvyklých forem HCM, které mohou být obtížně diagnostikovatelné echokardiograficky, např. apikální formy HCM. Oproti echokardiografii je senzitivnější v diagnostice hrotových aneurysmat, jež se vyskytují asi u 2 % nemocných. Velmi užitečná je možnost tkáňové charakteristiky pomocí kontrastního vyšetření gadoliniem, kdy pozdní sycení myokardu (tzv. late gadolinium enhancement – LGE) souvisí s přítomnost intramyokardiální fibrózy. V současnosti je zkoumán vztah mezi přítomností a rozsahem LGE a rizikem náhlé smrti u HCM (31). Endomyokardiální biopsie nemá v diagnostice HCM významné postavení. Je zatížena nezanedbatelným množstvím závažných komplikací a obvykle nepřináší u dospělých pacientů žádnou klinicky významnou informaci pro diagnózu ani terapii.

Ačkoli jsou základem diagnostiky HCM výše uvedené zobrazovací metody, podezření může vzniknout již na základě fyzikálního vyšetření a EKG. Fyzikální vyšetření může být u pacienta s HCM zcela normální, což je pravděpodobně jednou z příčin nízkého záchytu HCM v primární péči při předpokládané relativně vysoké prevalenci onemocnění. Mohou být přítomny nálezy reflektující přítomnost dynamické obstrukce ve výtokovém traktu levé komory srdeční (LVOT). Pulz na velkých tepnách může být rázný a bifidní s midsystolickým poklesem. Klasickým poslechovým nálezem dynamické obstrukce LVOT je crescendo-decrescendový systolický šelest, slyšitelný vlevo od sterna, s maximální intenzitou uprostřed a ke konci systoly. Na rozdíl od aortální stenózy se tento šelest nepropaguje do karotid. Intenzitu šelestu lze zvýraznit manévry, kterými snižujeme náplň LK –Valsalvův manévr či změna tělesné polohy z dřepu do stoje. Kromě tohoto typického šelestu může být patrný systolický šelest na srdečním hrotu způsobený mitrální regurgitací. Jen u 5 % jedinců s HCM je EKG křivka normální, u naprosté většiny nemocných je EKG patologické. Voltážové známky hypertrofie LK se vyskytují u více než tří čtvrtin pacientů. Často jsou doprovázené výraznými inverzemi vln T, zvláště vyjádřenými u apikální formy HCM. Patologické kmity Q, imitující stav po infarktu myokardu, se mohou objevit v jakémkoli svodu a jejich podkladem je nejspíše abnormální aktivace komorového septa. Častá je absence pozitivních kmitů R nebo porucha jejich gradientu v hrudních svodech. Mohou být přítomny poruchy nitrokomorového vedení (blokády Tawarových ramének, levý přední hemiblok). Doslova jakákoliv patologie ST úseku a T vln je u HCM možná. Holterovské monitorování EKG prokazuje u pacientů s HCM vysokou incidenci supraventrikulárních tachykardií, komorových extrasystol i nesetrvalé komorové tachykardie.
Patofyziologie HCM je komplexní a zahrnuje diastolickou dysfunkci, myokardiální ischémii, obstrukci výtokového traktu levé komory (LVOT), mitrální regurgitaci, supraventrikulární a komorové arytmie. Porucha diastolické funkce LK je patrná u všech nemocných s HCM již v časné fázi onemocnění. S progresí diastolické dysfunkce, spojené s rozvojem intersticiální fibrózy, dochází ke zvyšování tlaku v levé síni a postkapilární plicní hypertenzi. Zvyšování tlaku v levé síni a její dilatace má za následek rozvoj supraventrikulárních arytmií, nejčastěji fibrilace síní. Ischemie myokardu u HCM vzniká v důsledku hypertrofie myokardu a mikrovaskulární dysfunkce spojené s redukcí sítě kapilár (18). Klidová či provokovaná obstrukce LVOT je přítomná u přibližně dvou třetin nemocných s HCM a má řadu patofyziologických důsledků. Zvýšením afterloadu stoupá tlak v LK, což podporuje jak ischémii myokardu, tak zhoršení diastolických vlastností, a je též podkladem mitrální regurgitace, která dále zvyšuje tlak v levé síni. Při významné obstrukci dochází ke snížení srdečního výdeje. Podkladem obstrukce LVOT je dopředný pohyb jednoho či obou cípů mitrální chlopně v systole, tzv. SAM (systolic anterior motion), směrem k hypertrofickému bazálnímu septu, čímž dochází k zúžení LVOT. Mezi faktory podporující vznik SAM patří jak hemodynamické síly (akcelerace proudění krve kolem bazálního septa, doslova nasávající mitrální cípy do výtokového traktu), tak komplexní abnormalita celého aparátu mitrální chlopně. Charakteristický pro HCM je dopředný posun papilárních svalů a elongace cípů mitrální chlopně, zejména předního. Část pacientů s HCM má obstrukci midventrikulárně, v důsledku hypertrofie papilárních svalů nebo abnormální inzerce papilárního svalu přímo na přední cíp mitrální chlopně. Obstrukce LVOT je dynamická a mění se podle preloadu, afterloadu a kontraktility. U třetiny HCM pacientů je přítomna v klidu, u další třetiny pacientů je přítomna jen po provokaci (tzv. latentní obstrukce). Obstrukce LVOT je spojena s vyšším rizikem náhlé smrti a lze ji efektivně léčit septální redukční terapií. Alkoholová septální ablace účinně snižuje obstrukci LVOT i symptomatologii pacientů a je považována za bezpečný výkon za předpokladu provedení ve specializovaných centrech (32,33). Méně indikovanou alternativou v ČR je chirurgická myektomie.
Genetické studie z 80. a 90. let minulého století ukázaly, že příčinou hypertrofické kardiomyopatie jsou mutace genů pro proteiny sarkomery. V roce 1990 byla identifikována první jednoznačně kauzální mutace v genu pro těžký řetězec beta-myosinu (MYH7) (34). Během následující dekády byly identifikovány mutace v dalších genech pro sarkomerické proteiny (Tab. 4). Hypertrofická kardiomyopatie je onemocnění s vysokou fenotypovou a genotypovou variabilitou. Dosud bylo popsáno vice než 2000 mutací v 15 či více genech asociovaných s HCM (35,36). Kvůli genotypové heterogenitě je genetické vyšetření za použití konvenčních metod u HCM časově i finančně velmi náročné. Sekvenování nové generace (NGS) umožňuje analýzu velkého množství genů s podobnou přesností jako konvenční metody za významné úspory času i finančních prostředků. Díky stále větší dostupnosti NGS můžeme nyní získat genotyp od všech pacientů. Genetický screening velkého počtu genů má za následek časté nálezy tzv. variant nejasného významu (VUS) (19) a činí interpretaci genetického vyšetření velmi komplikovanou. Ideálním klinickým scénářem je identifikace jasně patogenní mutace u probanda, s následným cíleným kaskádovitým genetickým screeningem příbuzných, pátrajícím již po konkrétní mutaci. Kvůli nekompletní penetranci a variabilní expresivitě, která je pro HCM charakteristická, nemusí nález zděděné mutace na úrovni DNA znamenat rozvoj onemocnění se 100% pravděpodobností.
K manifestaci HCM může dojít kdykoli během života. Většina pacientů má mírné či žádné symptomy a diagnóza HCM je učiněna náhodně, např. při preventivní prohlídce nebo v rámci familiárního screeningu. Zásadními klinickými diagnostickými prostředky proto zůstávají i) pečlivý odběr rodinné a osobní anamnézy, ii) fyzikální vyšetření, iii) EKG vyšetření (vše dostupné v primární péči) a iv) echokardiografické vyšetření. Po stanovení diagnózy by měl být pacient referován do kardiocentra a doplněna další výše uvedená specializovaná vyšetření a riziková stratifikace náhlé smrti a případná indikace k septální ablaci.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
MUDr. Cyril Štěchovský
Kardiologická klinika 2. LF UK a Fakultní nemocnice v Motole
V Úvalu 84, Praha 15006
e-mail: stechovsky@gmail.com
tel.: 224434900
Zdroje
- Cooper LT, Knowlton KU. Myocarditis. In: Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli GF, Braunwald E eds. Braunwald‘s Heart Disease: A Textbook of Cardiovascular Medicine (11th ed). 2018 ISBN: 978-0-323-46342-3.
- Caforio AL, Pankuweit S, Arbustini E, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34 : 2636-2648.
- Kuethe F, Franz M, Jung C, et al. Outcome predictors in dilated cardiomyopathy or myocarditis. Eur J Clin Invest 2017; 47(7): 513-523.
- Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J Am Coll Cardiol 2009; 53(17): 1475-1487.
- Cooper LT, Baughman KL, Feldman AM, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Eur Heart J 2007; 28(24): 3076-3093.
- Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation 2003; 107(6): 857-863.
- Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J 2009; 30(16): 1995-2002.
- Moustafa A, Xie C, Kirkness E, et al. The blood DNA virome in 8,000 humans. PLoS Pathog 2017; 13(3): e1006292.
- Linhartová K, Hubáček P, Zemánek D, Kodetová D, Zajac M, Setina M, Veselka J. Presence of the viral genome in the myocardial tissue of patients without clinical suspicion of myocarditis. Cardiovasc Pathol 2013; 22(1): 113-114.
- Koepsell SA, Anderson DA, Radio SJ. Parvovirus B19 is a bystander in adult myocarditis. Cardiovasc Pathol 2012; 21(6): 476-481.
- Mason JW, O‘Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, Moon TE. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med 1995; 333(5): 269-275.
- Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005; 112(13): 1965-1970.
- Escher F, Kühl U, Lassner D, et al. Long-term outcome of patients with virus-negative chronic myocarditis or inflammatory cardiomyopathy after immunosuppressive therapy. Clin Res Cardiol 2016; 105(12): 1011-1020.
- Harmon KG, Drezner JA, Maleszewski JJ, et al. Pathogeneses of sudden cardiac death in national collegiate athletic association athletes. Circ Arrhythm Electrophysiol 2014; 7(2): 198-204.
- Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015; 116(12): 1887-1906.
- Singh BK, Pillai KK, Kohli K, Haque SE. Classification and Definitions of Cardiomyopathies. In: Veselka J ed. Cardiomyopathies, From Basic Research to Clinical Management. 2012 ISBN: 978-953-307-834-2.
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29(2): 270-276.
- Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet 2017; 389; 1253–1267.
- Bonaventura J., et al. The utility of the Mayo Score for predicting the yield of genetic testing in patients with hypertrophic cardiomyopathy. Arch Med Sci 2019; 15(3): 614-649.
- Hasselberg NE, Haland TF, Saberniak J, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J 2018; 39 : 853-860.
- Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016; 37(23): 1850-1858.
- Sliwa K, Hilfiker-Kleiner D, Petrie MC, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail 2010; 12(8): 767-778.
- Halliday BP, Wassall R, Lota AS, et al. Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED-HF): an open-label, pilot, randomised trial. Lancet 2019; 393 : 61-73.
- Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010; 31(7): 806-814.
- Waldmann V, Bougouin W, Karam N, et al. Characteristics and clinical assessment of unexplained sudden cardiac arrest in the real-world setting: focus on idiopathic ventricular fibrillation. Eur Heart J 2018; 39(21): 1981–1987.
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J 2014; 35 : 2733–2779.
- Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107 : 2227–2232.
- Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden Deaths in Young Competitive Athletes: Analysis of 1866 Deaths in the United States, 1980-2006. Circulation 2009; 119 : 1085–1092.
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65 : 1249–1254.
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 124(24): 783-831.
- Nagueh SF, Bierig SM, Budoff MJ, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: Endorsed by the American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2011; 24(5): 473-498.
- Veselka J, Jensen MK, Liebregts M, et al. Long-term clinical outcome after alcohol septal ablation for obstructive hypertrophic cardiomyopathy: results from the Euro-ASA registry. Eur Heart J 2016; 37(19): 1517-1523.
- Veselka J, Tomašov P, Januška J, Krejčí J, Adlová R. Obstruction after alcohol septal ablation is associated with cardiovascular mortality events. Heart 2016; 102 : 1793–1796.
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990; 62(5): 999-1006.
- Maron BJ, Maron MS, Semsarian C. Genetics of Hypertrophic Cardiomyopathy After 20 Years: Clinical Perspectives. J Am Coll Cardiol 2012; 60 : 705–715.
- Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015; 17(11): 880-888.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2019 Číslo 4
Najčítanejšie v tomto čísle
- Myokarditida a kardiomyopatie z pohledu kardiologa
- Fumarát hydratáza deficientní karcinom z renálních buněk a jemu podobný karcinom z renálních buněk: Komparativní studie 23 geneticky testovaných případů
- Inflamatorní myofibroblastický tumor dělohy – kazuistika
- Praktický přístup k pitvě srdce s vrozenou srdeční vadou