
Histopatologické principy vyšetření intersticiálních plicních procesů
Histopathology of interstitial lung diseases
Diffuse interstitial lung disorders include more than 200 different syndroms affecting the space between epithelial basal membrane and endothelial cells. Histopathological investigation of the lung tissue is one of the crucial parts of the multidisciplinary team approach for the investigation of these disorders. The aim of this review is a brief characterization of the pattern of the main subtypes of lung tissue damage.
Keywords:
interstitial – pulmonary – fibrosis – histopathology
Autoři:
Radoslav Matěj 1,2; Markéta Nová 3; Helena Hornychová 3
Působiště autorů:
Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
1; Ústav patologie 1. LF UK a VFN
2; Fingerlandův ústav patologie, Fakultní nemocnice a Lékařská fakulta Karlovy univerzity, Hradec Králové
3
Vyšlo v časopise:
Čes.-slov. Patol., 52, 2016, No. 2, p. 76-84
Kategorie:
Přehledové články - Interscitiální plicní procesy
Souhrn
Difúzní onemocnění plicního parenchymu zahrnují více než 200 různých syndromů s postižením prostoru mezi bazální membránou epitelové výstelky alveolů a endoteliemi cév. Histopatologické vyšetření plicní tkáně je jednou ze zásadních součástí multidisciplinárního přístupu v diagnostice těchto onemocnění. Účelem přehledného sdělení je charakteristika jednotlivých podtypů intersticiálních plicních procesů podle jejich převažujícího obrazu poškození plicní tkáně.
Klíčová slova:
intersticiální – plicní – fibróza – histopatologie
Difúzní onemocnění plicního parenchymu (DPLD) zahrnují více než 200 různých syndromů s postižením prostoru mezi bazální membránou epitelové výstelky alveolů a endoteliemi cév. Z hlediska morfologického vyšetřování je možné intersticiální plicní procesy (IPP) diagnostikovat z různých vzorků plicní tkáně, výtěžnost je však limitována technikou odběru a množstvím odebrané tkáně. Z tabulky 1 je zřejmé, že největší diagnostickou výtěžnost má správně provedená chirurgická plicní biopsie, ale ani v tomto případě nelze dosáhnout stoprocentní diagnostické úspěšnosti. Z hlediska histopatologického přínosu pro diagnostiku IPP je video-asociovaná torakoskopická biopsie (VATS) v 92 – 95 % ve shodě s otevřenou plicní biopsií z minitorakotomie dle Klassena. Existují ale některé histologické artefakty, které jsou častěji přítomny v biopsiích odebraných VATS. Zejména jde o častější oblasti recentního krvácení či přítomnosti neutrofilních granulocytů mimo cévní lumina (1-3).

Transbronchiální biopsie (TBB) má přínos k diagnostice IPP sice významný, ale omezený. Její význam stoupá, použije-li se modifikovaná transbronchiální kryobiopsie, kdy za použití nízkých teplot dojde pomocí kryosondy k odběru větší části plicního parenchymu. Tato metoda umožní získávat dostatečné množství tkáně pro diagnostiku IPP i z periferních oblastí, zatím však TBB kryobiopsie není zavedena do rutinní praxe na všech pracovištích.
Z hlediska histomorfologického hodnocení dochází při odebírání a zpracování vzorků získaných TBB k velkému množství nespecifických změn či dokonce jednoznačných artefaktů, které mohou někdy i zásadně měnit interpretaci tkáňových změn. V tabulce 2 je přehled těch nejčastějších a nejzásadnějších. U transbronchiální kryobiopsie však většina arteficiálních změn patrná není, proto jde o metodu, která bude hrát čím dál významnější roli v diagnostice IPP (4).

Asi nejdůležitějším artefaktem je komprese plicní tkáně v průběhu odběru, kdy takto mechanicky destruovaná tkáň může stištěním alveolárních sept a jejich relativní hypercelularitou simulovat intersticiální pneumonii. Rovněž rozdrcením jader zánětlivých elementů může docházet k obdobným artefaktům, které pozorujeme např. u malobuněčného karcinomu. Kompresí plicního parenchymu mohou také vzniknout různě velké bublinky vzduchu, jež mohou simulovat postižení exogenní lipoidní pneumonií. Povětšinou je lze odlišit (zejména vzhledem k nepřítomnosti obrovskobuněčné reakce), nicméně někteří méně zkušení patologové mohou být tímto obrazem zavedeni na scestí špatné diagnózy (3,5).
Dalšími změnami v souvislosti s odběrem mohou být oblasti recentního krvácení, které v některých specifických případech mohou dokonale imitovat až obraz akutní alveolární hemoragie. Poškození cévních stěn někdy může připomínat organizující se trombotizace v cévních luminech. Periferní odběry, které obsahují i části pleury s mezoteliálním krytem, mohou často napodobovat hyperplázii pneumocytů a tím budit dojem intersticiální fibrózy (6).
Klíčovým krokem pro správnou diagnostiku je rovněž správné zacházení s chirurgickou plicní biopsií. Existuje mnoho různých přístupů jak nakládat s materiálem získaným chirurgickou plicní biopsií. V každém případě je nutné zajistit úseky plicní tkáně pro kultivaci infekčních agens, výrazně navíc stoupá potřeba zajištění dostatečného množství tkáně pro případné následné genetické či molekulárně biologické analýzy. Nejdůležitějším však stále zůstává správné zacházení s materiálem pro účely vyšetření histomorfologického. Z hlediska použití fixativ připadají v úvahu různé varianty, nicméně stále platí, že ideálním standardním fixativem v histomorfologické diagnostice je pufrovaný formalín. Způsoby fixace jsou různé, záleží rovněž na zkušenostech a zvyklostech pracoviště. Na některých se používá metody napuštění fixativa do bronchiálního stromu, postupu, který často pomůže lépe objasnit strukturu plicního parenchymu, nicméně může být příčinou nevítaných artefaktů (zejména „emfyzematózních změn“ a „edému“) (7).
Zásadní je zdůraznit, že pouze úzká spolupráce a výměna informací mezi diagnostikujícím histomorfologem a ošetřujícím lékařem v korelaci s daty z radiodiagnostického vyšetření a výsledky dalších paraklinických metod, dostatečné množství řezů z různých úrovní odebrané tkáně a užití speciálních metodik vedou k maximalizaci diagnostické výtěžnosti vzorků plicního parenchymu, minimalizují procento nevýtěžných vzorků a významně snižují riziko zavádějící či dokonce mylné diagnózy. Diagnostikující histopatolog by měl být aktivním členem multidisciplinárního týmu (8).
Úlohou morfologického vyšetření je objektivně zhodnotit změny v odebrané tkáni. Základem hodnocení je rozpoznání postižení různých kompartmentů plicního parenchymu, kdy je třeba syntézou jednotlivých typů postižení dojít k diferenciálně diagnostickému závěru. Zjednodušeně se dá říci, že je třeba posoudit, KDE jsou změny lokalizovány, CO je jejich podstatou, KOLIK změn je v parenchymu a JAK jsou změny staré. Trendem poslední doby je určení dominující změny, ke které jsou přidruženy změny další, méně vyjádřené. Na základě pečlivé analýzy obrazu změn, jejich zastoupení a rozsahu lze dojít k histomorfologickému závěru, který obraz postižení plicního parenchymu nejvíce charakterizuje a je kompatibilní s klinickým nálezem (9).
Schémat k rozdělení lézí plicního parenchymu existuje celá řada, velmi populární je v poslední době schematické a didaktické rozdělení typů postižení dle Leslieho. Ten vychází z premisy, že změny postihující plicní intersticium mají pouze omezený repertoár histopatologických obrazů a popisuje šest základních typů histopatologického obrazu IPP (5).
Kombinace dominujícího histopatologického obrazu a obrazů vedlejších potom bývá specifická pro jednotlivé jednotky postižení plicního intersticia. V této klasifikaci je na prvním místě akutní plicní postižení (ALI), druhým obrazem je fibróza plicního parenchymu, třetím buněčná infiltrace plicní tkáně. Čtvrtý typ obrazu je charakterizován vyplněním alveolárních prostorů, pátý obraz reprezentují různé typy uzlovitých lézí. Poslední typ je vágně definovaný obraz „minimálních změn“, či „téměř normální plíce“ kam spadá postižení malých dýchacích cest, postižení cévních stěn a další léze, u nichž nenacházíme v přehledném zvětšení žádnou nápadnější změnu (3,5).
Obraz akutního intersticiálního plicního poškození (ALI)
Koncept akutního poškození plicního parenchymu je znám již řadu let, první autorkou byla Katzensteinová, která vycházela z poznatku, že plíce má pouze omezené spektrum reakcí na celou řadu různých akutních poškození. Dva nejčastější histopatologické obrazy spojené s reparací akutního poškození alveolů jsou obraz organizujícího se difúzního alveolárního poškození (DAD) a obraz organizující se pneumonie (OP). Pro oba tyto hlavní histopatologické obrazy je typická přítomnost fibroblastických proliferací stejného stáří (1).
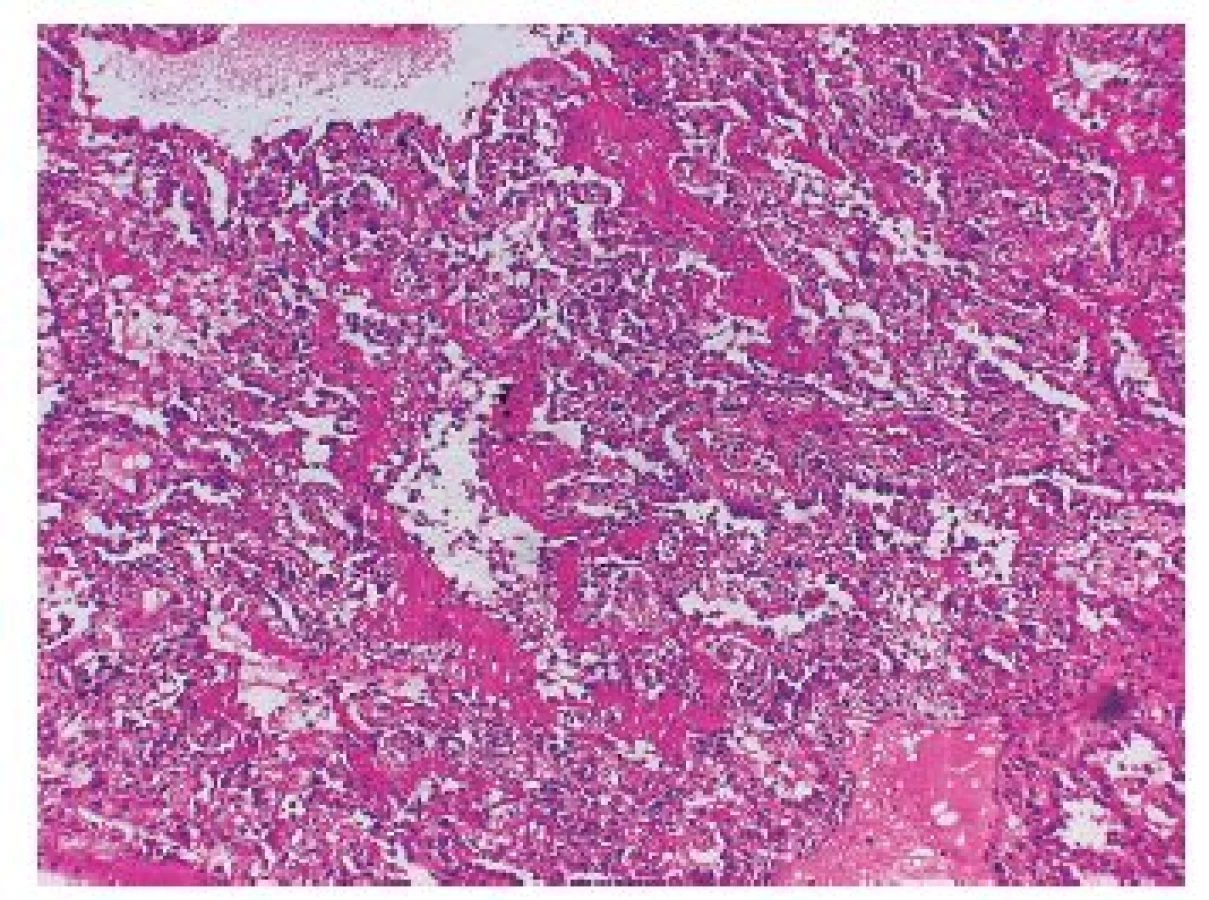
Histopatologický obraz DAD povětšinou koreluje s klinickou manifestací akutního respiračního selhání (ARDS). DAD znamená, že jsou poškozeny prakticky všechny části alveolů (epitelie, endotel i intersticiální stroma), slovo „difúzní“ však neznamená, že jsou vždy poškozeny všechny plicní úseky. Znamená, že DAD může, ale nemusí postihovat většinu plicního parenchymu, existují totiž i fokální formy DAD. Typické případy těžkého obrazu DAD jsou však zpravidla „difúzní“ a skutečně postihují většinu plicního parenchymu (obr. 1). Kromě největšího a nejzávažnějšího postižení ve smyslu DAD zpravidla bývají ještě vyjádřeny různé další znaky, které pak mohou přesněji charakterizovat klinické procesy asociované s tímto typem poškození plicního parenchymu (3,5).
Naproti tomu typickým histologickým znakem OP je polypoidní fibromyxoidní tkáň v bronchiolárních luminech, v alveolárních duktech i v distálních alveolech, která často tvoří poměrně dlouhé protáhlé masy. Intraluminální či intraalveolární polypoidní shluk nezralé fibrotické tkáně skládající se z fibroblastů a myofibroblastů se nazývá Massonovo tělísko (polyp) a předchozí studie prokázaly, že tato struktura může nejen progredovat a obliterovat alveolární prostory, ale může se propagovat i do interalveolárních sept i do malých dýchacích cest. Je tedy zřejmé, že tento typ postižení není selektivně lokalizován pouze intraalveolárně či intraluminálně a/nebo v intersticiálních prostorech, ale povětšinou postihuje oba tyto kompartmenty (obr. 2).

DAD a OP jsou poměrně snadno histopatologicky rozlišitelné, rovněž jejich klinický obraz je zcela rozdílný. V případě DAD jde obvykle klinicky o akutní, fulminantně probíhající onemocnění se špatnou prognózou, OP je naopak subakutní onemocnění, jehož prognóza může být i velmi příznivá.
Typický obraz DAD má dvě fáze: akutní a organizující se. Akutní neboli exsudativní fáze se objevuje během několika dní po traumatu a je charakterizována difuzním intersticiálním, intraalveolárním a septálním edémem a tvorbou hyalinních membrán v alveolárních duktech a alveolárních prostorech, v okolních cévách jsou čerstvé tromby, mnohdy zcela obliterující lumen. Později, zhruba za dva týdny, v organizující se fázi DAD, dochází k produkci nezralé intersticiální fibrotizace. Zřetelná je hyperplázie hyperregenerujících pneumocytů II. typu, které ztlušťují alveolární septa, patrné jsou rekanalizace trombů v okolních cévách.
Fibroblastické polypovité polštářky v organizující se pneumonii mohou obsahovat různá množství zánětlivé celulizace, někdy v nich nacházíme i obrovskobuněčné mnohojaderné elementy či části aspirované potravy. Často obraz OP pozorujeme v okolí granulomů, nekróz, tumorů, abscesů či vaskulitidy. Ve vzácnějších případech lze najít i maligní elementy přímo asociované s intraalveolární fibromyxoidní tkání (10).
Obraz OP bývá někdy minoritní komponentou jiných nemocí, které se projevují odlišným histopatologickým obrazem; často bývá přítomen při hypersenzitivní pneumonitidě (HP), nespecifické intersticiální pneumonii (NSIP), eozinofilní pneumonii (EP) či plicní histiocytóze z Langerhansových buněk (PLCH). Idiopatická OP bez zřejmé souvislosti s jakýmkoli známým inzultem plicního parenchymu je v současné době uznávaná jako samostatná klinicko-patologická jednotka a je označována adjektivem kryptogenní (COP).
Pokud v histopatologickém vyšetření nacházíme známky DAD i OP současně, nejlepším termínem je akutní plicní trauma (ALI), které respektuje obě subvarianty. Průkaz DAD či OP v biopsiích získaných endoskopickými metodami bývá zpravidla obtížný, tyto případy bývají často diagnostikovány jako akutní plicní poškození v korelaci klinického a radiologického obrazu (11-13).
I některé chronické intersticiální plicní nemoci mají složku akutního postižení plicního parenchymu. V případě idiopatické plicní fibrózy (IPF) byl zaveden klinický termín tzv. akutní exacerbace, charakterizovaný histopatologicky superpozicí DAD na obraz fibrotizující léze. Povětšinou jde o zásadní, a ve velké většině, fatální komplikaci původního chronicky probíhajícího intersticiálního plicního onemocnění s velmi nepříznivou prognózou (3,14).
Obraz fibrózy intersticia
Fibrózou rozumíme depozita organizovaných kolagenních vláken, která se jeví jako eozinofilní materiál ve standardně barvených řezech, a lze ji ozřejmit vybranými histochemickými metodami. Obraz fibrotizace plicního parenchymu je často přítomen u různých klinických jednotek, nicméně některá specifická uspořádání bývají charakteristická pro jednotlivé subtypy intersticiálních plicních onemocnění. Fibróza může být převážně nodulární nebo lineární či retikulární, její distribuce rovněž může být rozdílná s akcentací v periferii či bazálních lokalizacích, někdy v úsecích apikálních, jindy může být distribuována zcela náhodně v celém plicním parenchymu. Identifikace fibrózy v bioptickém vzorku může být ovlivněna mnoha různými faktory, např. mechanickými artefakty. Pro správné ozřejmení stupně fibrózy a její distribuce je důležité důkladné vyšetřování vzorků užitím specifických histochemických metod, zejména Massonovým trichromem. Je třeba připomenout, že vzorky získané transbronchiálními biopsiemi zpravidla nejsou v diagnostice fibrotizujících změn dostatečné a informují pouze o nevelkém úseku bez možnosti posoudit distribuci fibrotických změn.
Z hlediska distribuce fibrotických změn a podtypů obrazu fibrózy rozeznáváme několik relativně specifických jednotek.
Retikulární fibrotizace
Tento typ fibrotizace je velmi častý a představuje významný diagnostický problém pro odečítajícího patologa. Z hlediska klinicko-patologické kolerace bývá s výhodou rozdělit fibrotizující IPP na onemocnění s převahou změn v periferních a bazálních oblastech a oddělit je od změn v apikálních úsecích plicního parenchymu.
- a. Retikulární fibróza s distribucí v periferních a bazálních oblastech plicního parenchymu je typická pro obvyklý typ intersticiální pneumonie (UIP, idiopatická plicní fibróza – IPF), jejíž hlavní znaky jsou shrnuty v následném přehledném sdělení (obr. 3).

Nejdůležitější je odlišení IPF-UIP od rovněž velmi častého postižení plicního parenchymu charakteru nespecifické intersticiální pneumonie (NSIP) (obr. 4), což je jeden z nejdůležitějších a nejobtížnějších úkolů histopatologa v diferenciálně diagnostické rozvaze intersticiálních plicních procesů. Porovnání zásadních diagnostických znaků těchto dvou jednotek je uvedeno v tabulce 3.


Rovněž velké množství systémových nemocí pojiva, např. typicky revmatoidní artritida, systémová sklerodermie, polymyozitida/dermatomyozitida či systémový lupus erythematosus, může být komplikováno obrazem difúzního fibrotizujícího postižení plicního parenchymu. Ačkoli distribuce fibrózy v rámci této skupiny onemocnění může být zcela neodlišitelná od fibrózy charakteristické pro IPF-UIP, některé klinické entity mají tendenci vytvářet i jiná fibrotizující depozita (např. revmatoidní uzly), která mohou přispět k přesnější diagnostice fibrotizujících procesů. Odlišení IPF-UIP od pneumokonióz může také být v některých případech problematické. Např. azbestóza v pozdním stadiu může výrazně imitovat IPF-UIP histologicky, zejména pokud nebyla nalezena azbestová tělíska v histologických řezech. Také smíšené pneumokoniózy mohou být velmi obtížně odlišitelné od obrazu UIP, vyšetření v polarizovaném světle však může napomoci ozřejmit pravý původ fibrotizace. V histologickém obraze HP nacházíme bronchocentrické lymfoidní intersticiální infiltráty a různě formované granulomy v intersticiu s přítomností kolísavého počtu obrovskobuněčných elementů. Avšak v pokročilých stadiích a po protizánětlivé a imunosupresivní léčbě se může HP rovněž projevovat retikulární intersticiální plicní fibrózou a její odlišení od IPF-UIP bývá také velmi obtížné. Intersticiální pneumonie s přítomností obrovskobuněčných elementů, známá jako onemocnění plic v souvislosti s tvrdými kovy, musí být rovněž odlišena od IPF-UIP. Přítomnost velkého počtu obrovskobuněčných elementů zpravidla vede ke správné diagnóze, v některých případech se však predominance intersticiální fibrózy může stát příčinou diferenciálně diagnostických problémů v souvislosti s IPF-UIP či NSIP. Difúzní poškození alveolů (DAD) může být rovněž zaměněno s obrazem UIP. V klasickém obraze DAD je samozřejmě dominantní organizace fibrinu v alveolárních prostorech, která je následována převážně fibrózou nezralého typu, nicméně v pozdějších fázích hojení dochází k fibrotizaci a součástí obrazu jsou i změny charakteristické pro UIP (tzv. voština). Pro odlišení jsou důležité klinické údaje, farmakologická anamnéza a zejména průběh a závažnost onemocnění. ARDS je charakterizován náhlým život ohrožujícím onemocněním, obvykle v souvislosti se systémovým zánětem či traumatem. Problém ovšem nastává při tzv. akutní exacerbaci IPF, kdy například na obraz UIP jsou superponovány změny charakteristické pro DAD (3,5,15-17).
- b. Nejdůležitější jednotky spadající do skupiny retikulární fibrózy s distribucí v apikálních úsecích plicního parenchymu jsou porovnány v tabulce 4.

Nejčastější jednotkou v této skupině je sarkoidóza. V časných stadiích je toto onemocnění charakterizováno non-nekrotizujícím granulomatózním zánětem, onemocnění však může progredovat do difúzní intersticiální fibrózy i se změnami, které jsou jen obtížně odlišitelné od IPF-UIP. Přesto v případě sarkoidózy nacházíme na rozdíl od UIP hrubší fibrotizaci, která může mít za následek vznik bronchiektázií a emfyzému, stejně jako vytváření poměrně velkých cystických prostor v plicním parenchymu. Další jednotkou spadající do této kategorie fibrotizujících změn v plicním parenchymu je plicní histiocytóza z Langerhansových buněk. Intersticiální fibróza asociovaná s ankylózující spondylitidou postihuje sice pouze 1 % pacientů s tímto onemocněním, histologický obraz však bývá typicky charakterizován právě tímto typem distribuce fibrotických změn (6,7,10).
Nodulární fibrotizace
Obraz fibrotizace nodulárního charakteru je charakteristický zejména pro pneumokoniózy asociované s expozicí minerálnímu prachu obsahujícímu křemičitany. Tento typ fibrotizace se dělí dle velikosti nodulů na dva subtypy. Jde o postižení malými noduly a postižení, které je charakterizováno většími úseky fibrotizace.
- a. Prototypem onemocnění, které se projevuje malými noduly, je silikóza, pneumokonióza asociovaná s inhalací solí křemičitých krystalů. Jde o typické onemocnění spojené s pracovní expozicí anorganickým materiálům. Základní znak – silikotický uzel – je tvořen koncentrickými pruhy hustého kolagenního hyalinizovaného pojiva bez přítomnosti většího počtu buněk či zcela bezbuněčného. Depozita anorganických látek jsou zpravidla v periferii uzlu a jejich vlastnosti záleží na složení prachu. Často lze prachové částice ozřejmit polarizací.
- b. Splývající fibrotizace je definována nepravidelnými depozity kolagenu, která měří nejméně 2 cm. Prototypem tohoto typu postižení plicního parenchymu je progresivní masivní fibróza (PMF), devastující postižení plicní tkáně, které se může objevit kupříkladu v rámci uhlokopské plíce či Caplanova syndromu. Caplanův syndrom (revmatoidní pneumokonióza) je charakterizován masivními jizevnatými úseky v plicním parenchymu u horníků s revmatoidní artritidou. Masivní fibróza může být komplikací i původně silikotických uzlů, které mohou formovat poměrně velké agregáty. Tento typ postižení plicního parenchymu bývá rovněž pozorován i u dlouhodobých nitrožilních narkomanů postižených intravenózní talkózou, kdy částice talku nacházíme v trombózovaných cévních luminech a v okolní plicní tkáni s reakcí typu z cizích těles (3,7,18).
Lokalizovaná fibróza
Onemocnění, která jsou spojena s lokalizovanou fibrózou, je celá řada. U některých subtypů, např. u postradiační pneumonie, se formují spíše úseky apikálního postižení, u jiných je distribuce změn odlišná, s převahou ve střední části plicního parenchymu. Diagnostika těchto lézí bývá zpravidla dosti obtížná a morfologický nález musí být úzce korelován s klinikou i nálezem na zobrazovacích metodách. Pokud není nalezen žádný faktor, který by mohl být asociován s těmito změnami, je možné uzavřít diagnózu jako nespecifická fibrotizace nejistého původu. Charakteristickou podjednotkou s apikální distribucí je fibrotizace v apikálních úsecích horních segmentů dolního laloku, která se vyskytuje zpravidla u kuřáků, je velmi podobná postradiační pneumonii a pouze změny na cévách mohou vést ke správné diferenciální diagnóze. Zároveň musí být odlišeny kolagenní plaky na pohrudnici, ty bývají často asociovány s předchozí expozicí azbestu. Obdobným typem postižení se může projevovat rovněž reaktivace tuberkulózy, tu však doprovázejí i kavitace, kaseózní nekrózy a granulomatózní zánět. Příčinou obrazu lokalizované fibrózy mohou být i plicní infekce, především tuberkulóza. V lokalizovaných úsecích fibrotizace se mohou vyskytovat i sekundární změny. Nejčastějšími z nich jsou kalcifikace či osifikace.
Izolovanými depozity „fibrózy“ se projevuje též idiopatická pleuropulmonální (pleuroparenchymatózní) fibroelastóza (iPPFE). Jde o raritní IPP se současným postižením pleury a plicního intersticia. Fibrotické změny u PPFE mohou mít různou tíži a různou distribuci, často může být obraz asociován s jinými IPP. Zpravidla jsou postiženy maximálně subpleurální a centrilobulární oblasti, ale podrobné histopatologické analýzy nečetných popsaných případů ukazují, že distribuce změn je zpravidla disperzní. V pokročilých stádiích obraz PPFE je v přehledném barvení často neodlišitelný od obrazu UIP. Zcela zásadní pro správnou diagnózu je provést detekci elastických vláken, protože toto barvení umožní rozpoznat výraznou predominanci proliferace elastických vláken v projizvených úsecích plicní tkáně. Do obrazu postižení PPFE rovněž nepatří výraznější zánětlivá celulizace a výskyt granulomatózních formací (5,7,19).
Obraz výrazné zánětlivé infiltrace
Obraz postižení plicního parenchymu, kdy nejnápadnější změnou jsou různě rozsáhlé infiltráty smíšené zánětlivé celulizace, nacházíme u velkého množství jednotek. Kolísavý stupeň převážně chronické lymfoplazmocelulární zánětlivé celulizace je přítomen prakticky u všech intersticiálních plicních onemocnění, větší množství zánětu mimo voštinu je však například vylučujícím kritériem pro diagnózu UIP.
Dělení tohoto typu postižení je možné podle několika přístupů. Jedním z nich je možnost rozdělení specifických klinických jednotek podle distribuce zánětlivé celulizace. Akcentace smíšené zánětlivé celulizace může být v periferních úsecích plicního parenchymu zejména v subpleurálních oblastech, dále pak se může zánětlivá celulizace akumulovat peribronchiálně či perivaskulárně, v jiných případech nacházíme difúzní splývající infiltráty vyplňující interalveolární prostory. Z hlediska etiopatogenetického přístupu je rozdělení možné podle převažující komponenty zánětlivé celulizace. Přehled nejčastějších jednotek je shrnut v tabulce 5 (6,7).

Lymfocytární a plazmocelulární infiltrace
Klasickým představitelem jednotky, která je asociována s poměrně výraznou lymfocytární a plazmocelulární infiltrací, je HP. Distribuce zánětlivé celulizace je u HP proměnná stejně jako její intenzita. Ta velmi často závisí na předchozí léčbě imunosupresivními léčivy (zejména steroidy), která intenzitu a složení buněčného infiltrátu výrazně modifikují. Kromě převažující lymfo - a plazmocelulární zánětlivé celulizace nacházíme rovněž jen vágně formované granulomatózní formace, často bývají přítomny i jednotlivé obrovskobuněčné mnohojaderné elementy převážně typu z cizích těles, ale i Langhansova typu (obr. 5). Vždy je třeba důkladně pátrat v anamnéze pacienta po příčinné souvislosti s expozicí vyvolávajícímu antigenu (rozličné organické prachy) a korelovat s nálezem případných protilátek.

Rovněž pro skupinu intersticiálních plicních nemocí asociovaných se systémovými nemocemi pojiva jsou charakteristické různě intenzivní lymfoplazmocelulární infiltráty. Jejich lokalizace bývá rozdílná, poměrně nápadné jsou však změny na pleuře, kde kromě ztluštění nacházíme i známky různě aktivní nespecifické pleuritidy. Novou jednotkou postižení plicního parenchymu je intersticiální pneumonie s autoimunitními rysy (IPAF), která kombinuje histopatologický obraz různého postižení právě s akcentací lymfoplazmocelulární celulizace s různou distribucí (20).
Lymfocytární intersticiální pneumonie (LIP) je klinicko-patologická entita, kde smíšený lymfoplasmocelulární infiltrát tvoří i poměrně objemné agregáty v plicním parenchymu. V případech LIP je vždy nutné důkladně pátrat po klonalitě lymfoidního infiltrátu, protože podle nejnovějších poznatků je zřejmé, že pokud již LIP ve stadiu diagnózy není přímo asociována s lymfoproliferativní lézí, možnost vývoje hematologické malignity je velmi vysoká. Poměrně významnou složku obrazu buněčné NSIP tvoří rovněž smíšený zánětlivý infiltrát s převahou lymfoplazmocelulárních elementů. Obraz chronické zánětlivé infiltrace může doprovázet rovněž projevy toxicity asociované s některými chemickými látkami včetně léčiv. Je třeba zdůraznit, že nález intenzivnější lymfoplazmocelulární celulizace vždy vede k úvaze o možnosti postižení plicního parenchymu některým z lymfoproliferativních onemocnění, proto musíme vždy podrobně pátrat po složení zánětlivého infiltrátu a užitím specifických metod rozlišit, jde-li o proces monoklonální či polyklonální (5-7).
Infiltrace neutrofilními granulocyty
Nacházíme-li ve smíšeném zánětlivém infiltrátu větší frakci neutrofilních granulocytů, v první řadě je vždy nutné vyloučit infekční etiologii takového procesu, to včetně mykobakteriálních. Užitím série specializovaných metod lze často nalézt etiopatogenetická infekční agens, definitivní diagnózu však může přinést až kultivace s identifikací mikroorganismů. U některých případů systémových nemocí pojiva se na zánětlivé celulizaci rovněž mohou podílet častěji neutrofilní granulocyty, např. u revmatoidní artritidy či plicní granulomatózy s polyangiitidou. Neutrofilní granulocyty jsou rovněž často součástí postižení cévních stěn ve smyslu kapilaritidy u některých hemoragických syndromů (3,5,7).
Prominující fibrotizace s infiltrací lymfoplazmocelulárními elementy
Je-li součástí difúzní infiltrace lymfoplazmocelulárními elementy rovněž prominující fibrotizace, je nutné vždy pátrat po jednotkách asociovaných s fibrózou plicního parenchymu. Jak již bylo zmíněno, infiltráty zánětlivých elementů patří do obrazu většiny intersticiálních plicních procesů, záleží však na intenzitě a distribuci. Zatímco v případě buněčné NSIP může být zánětlivá celulizace i relativně nápadná, v případě obrazu IPF-UIP vede přítomnost výraznější zánětlivé celulizace spíše k diferenciálně diagnostické úvaze o možnosti jiné histopatologické diagnózy, na prvním místě je postižení v rámci systémové nemoci pojiva či HP.
Granulomatózní formace
Součástí smíšené zánětlivé celulizace mohou být i různé typy granulomatózních formací. Ať už se jedná o epiteloidní granulomy s přítomností centrální nekrózy či non-nekrotizující epiteloidní granulomy, vždy je třeba v první řadě vyloučit infekční etiologii těchto změn. Nález jen částečně ohraničených histiocytárních granulomů je charakteristický pro HP. Fibrotizované epiteloidní granulomy s přítomností různého počtu obrovskobuněčných mnohojaderných elementů v okolním smíšeném zánětlivém infiltrátu můžeme nalézt rovněž v časných stadiích sarkoidózy či beryliózy. V případě aspirace kromě granulomatózních formací nacházíme i obrovskobuněčné mnohojaderné elementy typu z cizích těles, v jejichž cytoplasmě jsou přítomny často zbytky aspirovaného materiálu, které je možno prokázat vyšetřením v polarizovaném světle či užitím některých specifických histochemických metodik. Po aspirovaném materiálu je nutné cíleně pátrat, což vyžaduje dostatečné množství řezů z různých rovin. Granulomatózní formace mohou být méně často nalezeny i v souvislosti s postižením plicního parenchymu jinými onemocněními (7,10,21).
Pleuritida
V případě smíšené zánětlivé celulizace, která je asociována s akutní či subakutní převážně serofibrinózní pleuritidou, je vždy nutné v první řadě pátrat po systémové nemoci pojiva, IPAF či jiné systémové autoimunitní nemoci, případně po možnosti polékového postižení (5,6,20).
Obraz intraalveolární exsudace a infiltrace
Intraalveolární infiltráty mohou být primárním procesem, ale často bývají sekundární změnou u jiných plicních onemocnění. Infiltráty mohou být buď bez výraznější přítomnosti buněk a obsahují významné množství eozinofilního materiálu či fibrinu, nebo mohou být buněčné a obsahovat různé typy zánětlivých elementů s převahou makrofágů či fibroblastických proliferací.
Klasickou jednotkou je plicní edém, který je povětšinou sekundární a souvisí se zvýšeným venózním tlakem, může však vznikat i přímo v souvislosti s poškozením alveolárních kapilár. Histomorfologicky je alveolární edém charakterizován akumulací jemné granulární eozinofilní hmoty v alveolárních prostorech. Jinou typickou jednotkou v rámci této skupiny je plicní alveolární proteinóza (PAP), vzácné onemocnění charakterizované akumulací proteinových, lipoproteinových a surfaktantu podobných materiálů v alveolárních prostorech (obr. 6). Součástí výplně alveolů jsou i poměrně objemná eozinofilní granula či opticky prázdné prostory po krystalech cholesterolu (tzv. cholesterolové hlatě), které usnadňují diferenciálně diagnostickou rozvahu proti prostému plicnímu edému. Příčinou PAP jsou buď vrozené, nebo získané poruchy tvorby a odbourávání surfaktantu. PAP může být idiopatická, v některých případech se však vyskytuje u imunodeficientních pacientů, někdy bývá asociována s nádorovými lézemi či lymfoproliferativními onemocněními a akutní masivní expozicí anorganických prachů (křemík, hliník).

Typickým představitelem infekčních agens, která se projevují alveolárním obrazem postižení plicního parenchymu, je Pneumocystis jirovecii, která typicky způsobuje pneumonii charakterizovanou intraalveolární akumulací exsudátu eozinofilního charakteru. Chronická zánětlivá celulizace v intersticiu je přítomna v kolísavém rozsahu, povětšinou je však jen nevelká. Intraalveolární materiál obsahuje drobné vakuoly, v nichž se dají speciálními metodami ozřejmit patogenní mikroorganismy (3,5-7,10).
Relativně klasickým obrazem spadajícím do kategorie intraalveolárních exsudátů a infiltrátů se projevuje i akutní pneumonie. V tomto případě v dilatovaných alveolárních prostorech nacházíme kolísavý stupeň smíšené zánětlivé celulizace s výraznou převahou neutrofilních granulocytů. Obdobným příkladem jsou další typy pneumonií asociovaných buď s eozinofilní pneumonií či endogenní/exogenní lipoidní pneumonií.
Obraz uzlů – granulomy a granulomatózní léze
Histomorfologický pojem granulom odpovídá různou měrou ohraničené nodulární lézi tvořené agregátem buněk. Podrobnější přehled granulomatózních plicních lézí je uveden v jiném přehledném článku v tomto čísle časopisu.
Obraz „minimálních změn“ ČI „TÉMĚŘ NORMÁLNÍ PLÍCE“
Někdy se stává, že vyšetřovaný materiál z chirurgické plicní biopsie obsahuje jen zcela minimální histologické změny i při signifikantních klinických a radiografických nálezech. V první řadě může jít samozřejmě o následek neadekvátního odběru plicní tkáně. Některé klinické jednotky však mohou být asociovány právě s tímto nálezem. Časný intersticiální edém v souvislosti se srdečním selháváním může mít za následek nevelké rozšíření interlobulárních sept či dilataci lymfatik. V oblasti chronických difúzních infiltrativních procesů je jednou z klasických jednotek, které mohou mít v chirurgické plicní biopsii výše popsaný nález „minimálních“ změn, PLCH v raných stadiích, zejména je-li její diagnostika spojena s velmi omezeným odběrem plicního parenchymu. Dalším zásadním faktorem ovlivňujícím histomorfologický obraz intersticiálních plicních procesů je léčba kortikosteroidy před diagnostickým odběrem. Může dojít k výraznému potlačení zánětlivých změn, v plicním parenchymu zůstávají pouze nevelké lymfoplazmocelulární perivaskulární infiltráty a hyperplázie pneumocytů II. typu. Obraz „minimálních změn“ bývá i při plicní hypertenzi a útlaku plicního parenchymu z extrapulmonálních příčin jako např. při obezitě či neuromuskulárních onemocněních. V tabulce 6 jsou nejčastější příčiny obrazu „minimálních změn“ shrnuty. Vždy je však třeba pečlivě analyzovat všechny kompartmenty plicního parenchymu, aby nedošlo k přehlédnutí onemocnění malých dýchacích cest či cévních struktur, vhodné je vždy doplnění speciálních histochemických metod (zejména průkaz elastických vláken k ozřejmení konstriktivní bronchiolitidy či vaskulitických změn) a imunohistochemických metod (CD1a či S100 protein k ozřejmení PLCH) (5-7,10).

Plicní hemoragie
Krvácení do plicního parenchymu je poměrně častým histologickým nálezem. Může jít o akutně vzniklý proces s extravazací erytrocytů nebo jsou nalezena depozita hemosiderinu jako známky staršího krvácení. Krvácení může být fokální nebo může difúzně postihovat celý plicní parenchym. Častá je velmi dramatická klinická manifestace, někdy ale může jít pouze o náhodný nález bez klinického korelátu. V diferenciálně diagnostické rozvaze je zásadní analýza klinických údajů, která může přispět k objasnění příčiny krvácení. V bioptickém vzorku je často obtížné rozeznat, jedná-li se o opravdové krvácení do plicního parenchymu, či jde o artefakt vzniklý v průběhu odběru. Ve vzorcích získaných TBB jsou arteficiální hemoragie velmi časté, obdobný problém se však týká i chirurgických plicních biopsií odebraných VATS. Platí, že akutní plicní krvácení nelze histopatologicky diagnostikovat, pokud nejsou k dispozici relevantní klinická data. Hemosiderin jako klíčový pigment staršího krvácení je nutné odlišit od ostatních typů pigmentů, které se vyskytují v plicích. Hemosiderin je tmavohnědý až zlatavý granulární pigment, zatímco „kuřácký“ pigment je světle špinavě hnědý (3,5,10).
ZÁVĚR
Histopatologická diagnostika IPP vyžaduje podrobnou evaluaci všech kompartmentů plicní tkáně. Její výtěžnost kromě zkušenosti vyšetřujícího patologa závisí na mnoha faktorech. Vzorky plicní tkáně by měly být vyšetřeny extenzivně, vždy je třeba uvážit možnost užití speciálních vyšetřovacích metod. Zcela zásadním aspektem je technika odběru, kdy nevhodně či suboptimálně odebraný vzorek může být naprosto limitující a bez znalostí komplexních údajů o pacientovi, způsobu a místě odběru může vést ke špatné diagnóze. Klíčovým faktorem je tedy úzká multidisciplinární spolupráce a diagnostika intersticiálních plicních procesů by měla VŽDY být souhrnným dílem spolupracujícího pneumologa, radiologa a patologa.
Na úplný závěr, pro všechny, co dočetli až sem, doplňujeme seznam nejužívanějších zkratek:
ALI – akutní plicní trauma, COP – kryptogenní organizující se pneumonie, DAD – difúzní alveolární poškození, DIP – deskvamativní intersticiální pneumonie, DLPD - difúzní onemocnění plicního parenchymu, HP – hypersenzitivní pneumonitida, IPAF – intersticiální pneumonie s autoimunitními rysy, IPF – idiopatická plicní fibróza, IPP – intersticiální plicní procesy, LIP – lymfocytární intersticiální pneumonie, NSIP – nespecifická intersticiální pneumonie, PLCH – plicní histiocytóza z Langerhansových buněk, PPFE - pleuropulmonální (pleuroparenchymatózní) fibroelastóza, RB-ILD – respirační bronchiolitida s intersticiálním plicním postižením, TBB – transbronchiální bronchoskopická biopsie, UIP – obvyklá intersticiální pneumonie
PODĚKOVÁNÍ
Poděkování patří všem naším kolegům (plicní kliniky, radiodiagnostická pracoviště, oddělení hrudní chirurgie), bez jejichž spolupráce by kvalitní histopatologická diagnostika intersticiálních plicních procesů nebyla možná. Práce vznikla za částečné podpory projektů PRVOUK P37/11 a P27/LF1/1, OPPK CZ.2.16/3.1.00/24509 a BBMRI LM2010004.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
doc. MUDr. Radoslav Matěj, Ph.D.
Oddělení patologie a molekulární medicíny
Thomayerova nemocnice
Vídeňská 800,
14059 Praha 4 - Krč,
e-mail: radoslav.matej@ftn.cz
tel.: +420 261083741
fax: +420 234333742
Zdroje
1. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157 : 1301–1315.
2. Leslie KO. Historical perspective. A pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(5): 513–519.
3. Vašáková M, Polák J, Matěj R. Intersticiální plicní procesy. Maxdorf, Praha; 2011.
4. Tomassetti S, Wells AU, Costabel U,et al. Bronchoscopic Lung Cryobiopsy Increases Diagnostic Confidence in the Multidisciplinary Diagnosis of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2016, in press.
5. Leslie KO, Wick MR. Practical pulmonary pathology (2nd ed). Philadelphia: Elsevier; 2011
6. Hasleton P, Flieder DB. Spencer´s Pathology of the Lung, Sixth Edition. Cambridge University Press; 2013.
7. Corrin, B, Nicholson, AG. Pathology of the Lungs. Third Edition. Churchil Livingstone Elsevier; 2011.
8. Maffessanti M, Dalpiaz G. Diffuse Lung Diseases. Clinical Features, Pathology, HRCT. Springer, 2006.
9. Jones KD, Urisman A. Histopathologic approach to the surgical lung biopsy in interstitial lung disease. Clin Chest Med 2012; 33(1): 27-40.
10. Popper HH. Interstitial lung diseases-can pathologists arrive at an etiology-based diagnosis? A critical update. Virchows Arch 2013; 462(1): 1-26.
11. Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax 2003; 58 : 143–148.
12. Karthikeyan D. High-resolution Computed Tomography of the Lungs. A Pattern Approach. Hodder Arnold; 2005.
13. Stern EJ, Swensen JS, Kanne JP. High-Resolution CT of the Chest. Wolters Kluwer, Lippincott Williams, Wilkins; 2010.
14. Cagle PT, Allen TC, Beasly MB. Diagnostic Pulmonary Pathology, Second Edition. Informa Healthcare; 2008.
15. Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6): 788-824.
16. Travis WD, Costabel U, Hansell DM et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188(6): 733-748.
17. Spagnolo P, Sverzellati N, Rossi G et al. Idiopathic pulmonary fibrosis: an update. Ann Med 2015; 47(1): 15-27.
18. Travis WD, Colby TV, Koss MN, et al. Non-neoplastic disorders of lower respiratory tract. Washington: Armed Forces Institute of Pathology; 2002.
19. Rosenbaum JN, Butt YM, Johnson KA, et al. Pleuroparenchymal fibroelastosis: a pattern of chronic lung injury. Hum Pathol 2015; 46(1): 137-144.
20. Fischer A, Antoniou KM, Brown KK et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015; 46(4): 976-987.
21. Castonguay MC, Ryu JH, Yi ES, Tazelaar HD. Granulomas and giant cells in hypersensitivity pneumonitis. Hum Pathol 2015; 46(4): 607-613.
Štítky
Patológia Súdne lekárstvo ToxikológiaČlánok vyšiel v časopise
Česko-slovenská patologie

2016 Číslo 2
Najčítanejšie v tomto čísle
- Diferenciální diagnostika granulomatózních procesů v plicích
- Intersticiální plicní onemocnění asociovaná s kouřením
- Idiopatická plicní fibróza - problematika multidisciplinární diagnostiky a léčby ve světle nových poznatků
- Histopatologické principy vyšetření intersticiálních plicních procesů