
Atrophodermia vermiculata u chlapce s Marfanovým syndromem
Atrophodermia Vermiculata in a Boy Suffering from Marfan’s Syndrome
Authors introduce a histologically confirmed case of atrophodermia vermiculata in a 10-year-old boy fulfilling some criteria for Marfan’s syndrome. They present an overview of opinions regarding clinical appearance, differential diagnosis, treatment and possible association with Marfan’s syndrome.
Key words:
atrophodermia vermiculata – diagnosis – differential diagnosis – Marfan’s syndrome
Autoři:
M. Hašková 11; H. Duchková 11; P. Arenberger 22; L. Pock 3
Působiště autorů:
Kožní sanatorium, Ústí nad Labem
vedoucí lékařka MUDr. Hana Duchková, DrSc.
1; Dermatovenerologická klinika, FNKV, Praha
přednosta prof. MUDr. Petr Arenberger, DrSc., MBA
2; Dermatohistopatologiocká laboratoř, Praha
vedoucí lékař doc. MUDr. Lumír Pock, CSc.
3
Vyšlo v časopise:
Čes-slov Derm, 88, 2013, No. 6, p. 288-291
Kategorie:
Kazuistiky
Souhrn
Autoři uvádí histologicky potvrzený vzácný případ kožního onemocnění – atrophodermia vermiculata. Prezentují přehled současných poznatků o klinických projevech, diferenciální diagnostice, léčbě a možném sdružení s Marfanovým syndromem. Demonstrují desetiletého chlapce s diagnózou atrophodermia vermiculata splňující některá kritéria Marfanova syndromu.
Klíčová slova:
atrophodermia vermiculata – diagnóza – diferenciální diagnóza – Marfanův syndrom
Atrophodermia vermiculata je kožní onemocnění řazené mezi genodermatózy. Začínaná mezi 5. až 12. rokem života. Je charakterizované ostře ohraničenými vkleslinami různých velikostí a tvarů na zarudlé spodině. Postiženy jsou tváře, někdy obočí, ušní lalůčky. Onemocnění má převážně symetrickou distribuci. Sdružení s různými syndromy je možné [2, 4, 11]. Překládáme případ našeho nemocného s kožními projevy sdružené s Marfanovým syndromem.
POPIS PŘÍPADU
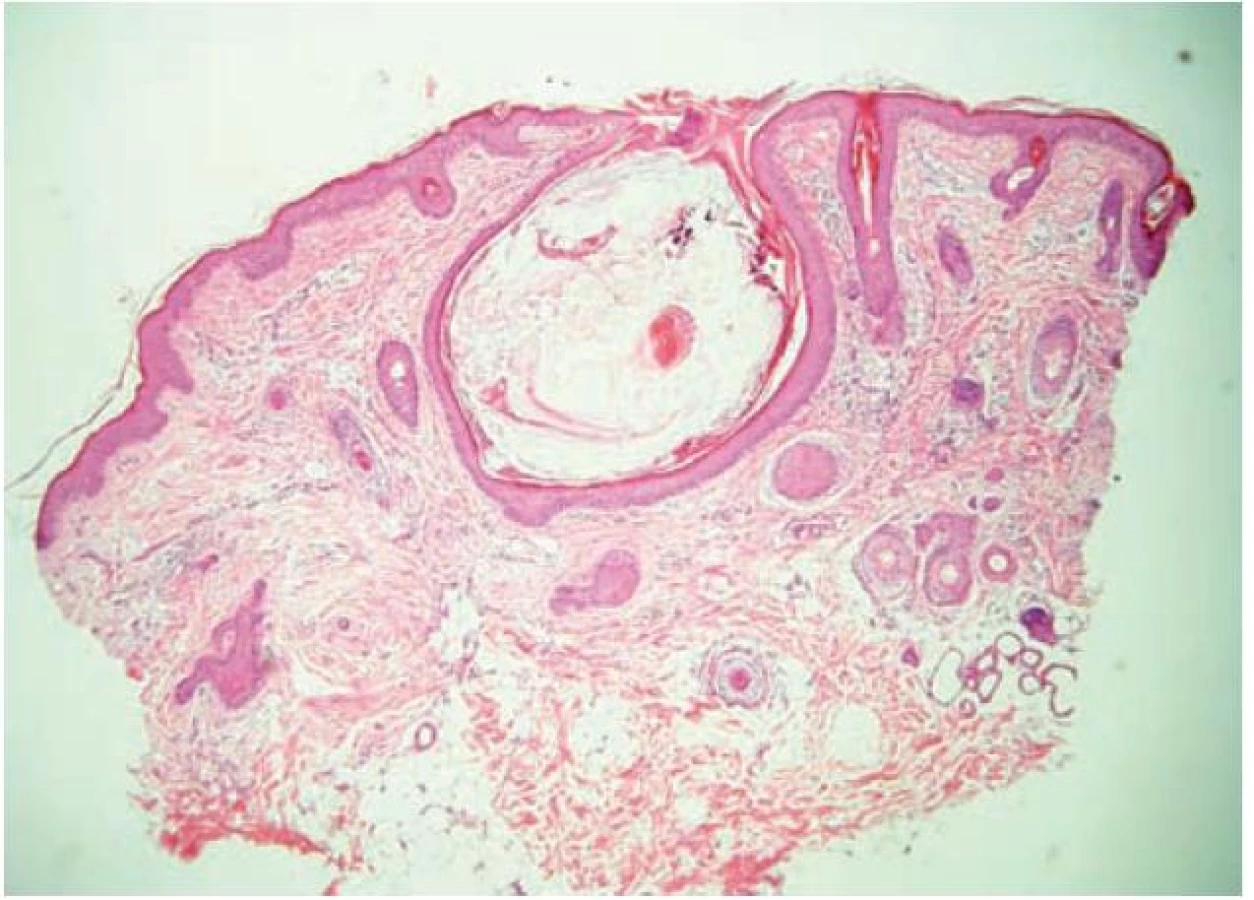
Poprvé se kožní změny objevily na tvářích a zevních třetinách obočí v pěti letech. Postupně docházelo k progresi. Při vyšetření byla kůže na tvářích a v obočí, méně na ušních lalůčcích, difuzně zarudlá s folikulárně vázanými papulami velikosti špendlíkové hlavičky. Nápadné byly ostře vyseknuté vklesliny nepravidelných tvarů, velikosti od 1–2 mm, hloubky až 1 mm, místy mřížkového vzhledu. Ojediněle byla patrna milia. V zevních třetinách obočí a na ušních lalůčcích byly podobné změny jako na tvářích (obr. 1a, b). Kůže na ostatních částech těla byla bez patologických změn. V útlém dětství se vyskytovaly patrně kožní příznaky atopické dermatitidy. Podobně tak u sestry nemocného, u tety Crohnova nemoc. Pacient pocházel z první gravidity matky, porod měl fyziologický průběh, v termínu 39 + 3, záhlavím. PH 3950/54 cm, novorozenec nebyl kříšen. Poporodní adaptace byla v v normě, ikterus byl slabý. Psychomotorický vývoj byl bez známek opoždění. Později byla zjištěna porucha psaní. Kostní věk 10letého chlapce odpovídal věku 13 let – výška byla 150 cm. Endokrinologické a antropometrické vyšetření konstatovalo souvislost vyššího vzrůstu s časnější iniciací puberty, byla prokázaná urychlená osifikace, byly zjištěny delší končetiny, naznačená skolióza páteře, arachnodaktylie, vpáčené sternum. Karyotyp byl v normě – 46, XY. Vyšetřující lékařka vzhledem k možné autozomálně recesivní dědičnosti upozornila na riziko vzniku kožního onemocnění u dalších sourozenců v poměru 1 : 4. Gen pro tuto afekci nebyl dosud popsán. Genealogické vyšetření neprokázalo u otce, matky ani u sourozenců příznaky onemocnění. Kontrolní vyšetření provedené v 11 letech věku prokázalo urychlený vzrůst – výška byla 172 cm (za rok nárůst o 22 cm), další prodlužování končetin, prstů (arachnodaktylie) a výrazněji vpáčený hrudník (obr. 2a, b), skoliózu, zkrácení levé dolní končetiny o 7 mm. Oční vyšetření ozřejmilo oboustrannou hypermetropii (sníženou zrakovou ostrost a absenci konvergentního souhybu), kardiologické vyšetření zjistilo lehkou dilataci aortálního bulbu svědčící pro Marfanův syndrom. Histologické vyšetření prokázalo v dilatovaném infundibulu vlasového folikulu velký hyperkeratotický čep tvořený jemným keratinem, v něm stočený velusový vlas s přítomností malého množství tkáňového detritu s nekrotickými polynukleáry – tento spojuje dva sousední vlasové folikuly. Epiteliální stěna folikulu je poměrně tenká, v okolí je mírná fibrotizace a perivaskulárně malé infiltráty lymfocytů s nečetnými histiocyty a plasmocyty. V jednom místě je i drobný granulom z cizích těles, reakce na keratin, pravděpodobně z perforovaného folikulu. Ložiskovitě je ve fibrotizované části koria patrná redukce až vymizení elastických vláken. Závěr histologického vyšetření: popsaný obraz je v souladu s diagnózou atrophodermia vermiculata (obr. 3).




DISKUSE
Atrophodermia vermiculata (označovaná také jako Honey-Comb atrophy, Folliculitis ulerythematosa, Atrophodermia ulerythematosa) patří do skupiny folikulárních keratóz. Kožní změny postihují primárně tváře, oblast před ušními boltci, obočí, někdy ušní lalůčky – oboustranně, méně často jednostranně. Mozaikovité vklesliny dělají dojem voštin včelího plástu („honey-com - -bed“) nebo dojem vyžrání od červů („worm eaten-like scars“). Onemocnění vzniká v dětském věku, většinou před pubertou, zřídka i v pozdějším věku, se sklonem k regresi [6]. V diferenciální diagnostice je nutno odlišit především choroby patřící do skupiny folikulárních keratóz. Keratosis pilaris rubra atrophicans faciei (Ulerythema ophryogenes). Keratosis pilaris rubra atrophicans se považuje za podtyp ulerythema ophryogenes, má podobný klinický i histologické obraz, liší se však lokalizací prvních projevů. Ulerythema ophryogenes začíná primárně v obočí, keratosis pilaris rubra atrophicans faciei začíná primárně na tvářích. Onemocnění jsou charakterizovaná přesně ohraničeným erytémem, malými keratotickými folikulárně vázanými papulemi na tvářích, čele, bradě a v preaurikulární oblasti. Někteří popisují dvojí druh změn podle toho, zdali je přítomná nebo nepřítomná zánětlivá reakce. Význam tohoto příznaku mnozí popírají. Drsnost kůže je jedním z popisovaných symptomů [7]. Postižení zevní třetiny obočí může vést ke vzniku jizvící alopecie. Nad extenzory končetin bývá keratosis pilaris. Onemocnění začínají v útlém dětství. Atrophodermia vermiculata se předchozí chorobě podobá především lokalizací na tvářích i obočí, přesně ohraničeným zarudnutím. Drsnost kůže není dominantní, vklesliny připomínající vykousání od červů je velmi typické. Keratosis pilaris nad extenzory se nevyskytuje často, podobně tak nedochází často k jizvící alopecii v obočí. Na rozdíl od keratosis pilaris atrophicans faciei vzniká atrophodermia v pozdějším věku – před pubertou, nebo i později.
V diferenciální diagnóze lze zvážit ještě Keratosis spinulosa decalvans (Siemens) postihující kštici, trup, dorza rukou, začínající v raném dětství a Atrophodermia maculosa varioliformis charakterizovaná výskytem mělkých drobných jizev bez zánětlivé reakce lokalizovaných na obličeji. Podobnost s atrophodermia vermiculata je mizivá.
Atrophodermia vermiculata se sdružuje se s Marfanovým, méně často s ROMBO a Nicolau-Balusovým syndromem. Marfanův syndrom je autosomálně-dominantní dědičné onemocnění postihující pojivovou tkáň kostry, oka (poruchy čočky, její subluxace) a kardiovaskulárního systému (postižení mitrální chlopně a aorty s možným vznikem jejího aneurysmatu). Postižení mají typický habitus s vysokou postavou, dlouhými tenkými končetinami a prsty (arachnodaktylie), trychtýřovitý hrudník, vpáčené sternum, skoliózu. ROMBO syndrome vykazuje mnohočetná milia, hypotrichózu, trichoepitheliomy, bazaliomy, periferní vazodilataci s cyanózou. Nicolausův-Balusův syndrom představuje erupce mnohočetných syringomů a milií. Keratosis pilaris rubra atrophicans faciei se vyskytuje u jiných syndromů – u Noonanova a „Wooly hair“ syndromu. Noonanův syndrom – osoby s malou postavou, nízkou inteligencí, s hypertrofickou kardiomyopatií, pulmonální stenózou. „Wooly Hair syndrome“ (syndrom kudrnatých vlasů) představují osoby s bílými, krátkými a kudrnatými vlasy, u nichž jsou pruhovité hyperkeratózy na dlaních a chodidlech. Atrophodermia vermiculata u těchto syndromů popsaná nebyla.
Atrophodermia vermiculata je z hlediska morfologie charakteristická, téměř nelze ji zaměnit za jiné choroby. Vzniká také v pozdějším věku než srovnatelné onemocnění keratosis pilaris rubra atrophicans faciei, vyskytuje se také u jiných syndromů. Jansen se dokonce domnívá, že se jedná o onemocnění „sui genesis“ [1, 2, 3, 4, 5, 7, 8, 10, 11].
Léčba je symptomatická. Lokálně se používají externa – emoliencia s ureou, kyselinou mléčnou, dále lokální kortikosteroidy a retinoidy. V celkové terapii lze podle závažnosti použít i isotretinoin [12]. Uspokojivé výsledky byly dosaženy s použitím pulzního laseru o vlnové délce 595 nm (pulsed dye laser) a intenzivního pulzního světla (IPL) [9]. Náš pacient v současné době prodělává léčbu intenzivním pulzním světlem, účinnost zatím nelze hodnotit. Ortoped plánuje symptomatickou chirurgickou léčbu – reparaci skoliózy a vpáčeného hrudníku.
ZÁVĚR
Atrophodermia vermiculata, vzácná genodermatóza, byla diagnostikována u chlapce, u něhož došlo postupně k rozvoji dalších patologických změn svědčících pro Marfanův syndrom. Vzhledem k tomu, že je pacient ohrožen různými komplikacemi, z nichž nejzávažnější je disekce aorty, je nutno zabezpečit celoživotní cílené kontroly na specializovaných pracovištích. Koordinaci sledování pacienta zajišťuje kožní oddělení.
Do redakce došlo dne 19. 9. 2013.
Adresa pro korespondenci:
MUDr. Marta Hašková
Kožní sanatorium, s. r. o.
Velká Hradební 47
400 11 Ústí nad Labem
haskova@koznisanatorium.cz
Zdroje
1. ARNOLD, A. W., BUECHNER, S. A. Keratosis pilaris and keratosis pilaris atrophicans faciei. J. Dtsch. Dermatol. Ges., 2006, 4, p. 319–323.
2. ASHINOF, R., JACOBSON, M. Rombo syndrome a second case report and review. J. Am. Acad. Dermatol., 1993, 28, p. 1011–1014.
3. BRAUN-FALCO et al. Disorders of keratinization. Dermatology, 2000, pp. 700–750.
4. DUPRÉ, A. et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus syndrome. Dermatologica, 1981, 162 (4), p. 281–286.
5. GONZALEZ, J. et al. Keratosis pilaris rubra and keratosis pilaris atrophicans faciei treated with pulsed dye laser: report of 10 cases. JEADV, 2011, 25, p. 710–714.
6. JANSEN, T., SANDER, C. A., ALTMEYER, P. Atrophodermia vermiculata: case report and review of the literature. JEADV, 2003, 17, p. 70–72.
7. KODET, O., GEMPERLOVÁ, V., ŠTORK, J., LACINA, L. Klinický případ: Drsné papuly v obličeji. Čes-slov Derm., 85, 2010, No. 4, p. 225–227.
8. KOLENIK, S. A., PEREZ, M. I. Atrophia maculosa varioliformis cutis. Report of two cases and review of the literature. J. Am. Acad. Dermatol., 1994, 30, p. 837–840.
9. KAUNE, K. M., HAAS, E. Succesful treatment of severe keratosis pilaris rubra with a 595 nm pulsed dye laser. Dermatol. Surg., 2009, 35, p. 1592–1595.
10. PIERINI, D. O., PIERINI, A. M. Keratosis pilaris arophicans faciei (ulerythema ophryogenes): a cutaneous marker in the Noohan syndrome. Br. J. Dermatol., 1979, 100, p. 409–414.
11. SIDWEL, R. U., HARPER, J. I. Vermiculate atrophoderma in a boy with Marfan syndrome. Br. J. Derm., 1999, 141, p. 750–751.
12. WEIGHTMAN, W. A. A case of atrophoderma vermiculatum responding to isotretinoin. Clin. Exp. Dermatol., 1998, 23, p. 889–810.
Štítky
Dermatológia Detská dermatológiaČlánok vyšiel v časopise
Česko-slovenská dermatologie

2013 Číslo 6
- Na český trh přichází biosimilar adalimumabu s prokázanou terapeutickou ekvivalencí
- Nehoňte nemocné s mMCC od čerta k ďáblu!
- První a jediná schválená imunoterapie vzácného agresivního karcinomu kůže
- Screening malignit u pacientů s dermatomyozitidou
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
Najčítanejšie v tomto čísle
- Granuloma anulare
- Onemocnění ruka-noha-ústa
- Symetrické léze – superficiální maligní melanomy s nízkými hodnotami ABCD skóre
- Klinický případ: Bělavé makuly na zádech