
Histiocytární choroby
Histiocytic disorders
Histiocytic disorders occur sporadically and may thus be considered as rare diseases. The most frequent serious histiocytic disorder of adulthood is the Langerhans cell histiocytosis. Juvenile xanthogranuloma is associated with foamy histiocytes and affects patients in early childhood. Erdheim-Chester disease, on the other hand, is a disease of adulthood. Foamy macrophages usually infiltrate lower extremities as well as retroperitoneum and mediastinum and are the cause of fibrosis. Both Erdheim-Chester disease and Langerhans cell histiocytosis sometimes infiltrate hypothalamus and pituitary stalk and diabetes insipidus is thus the first sign of the disease. Dendritic cell sarcoma occurs very rarely and is, unlike the former two diseases, resistant to chemotherapy.
Key words:
histiocytosis – Langerhans cell histiocytosis – juvenile xanthogranuloma – Erdheim-Chester disease – dendritic cell sarcoma
Autoři:
Z. Adam; M. Krejčí; L. Pour
Působiště autorů:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Vorlíček, CSc.
Vyšlo v časopise:
Vnitř Lék 2009; 55(Suppl 1)(Supplementum 1): 109-124
Souhrn
Histiocytární choroby se vyskytují velmi ojediněle, lze je tedy zařadit do vzácných chorob. V dospělosti se nejčastěji ze všech závažných histiocytárních chorob vyskytuje histiocytóza z Langerhansových buněk. Juvenilní xantogranulomatóza se odvozuje z pěnitých histiocytů. Postihuje pacienty v raném dětském věku. Erdheimova-Chesterova nemoc je naopak typickou chorobou věku dospělého. Pěnité makrofágy obvykle infiltrují nejenom kostní dřeň dlouhých kostí dolních končetin, ale také retroperitoneu a mediastinu a způsobují fibrózu. Jak Erdheimova-Chesterova choroba, tak histiocytóza z Langerhansových buněk někdy infiltruje hypothalamus a stopku hypofýzy, takže první známkou nemoci nejen v dětství, ale také v dospělosti je diabetes insipidus. Zcela vzácně se vyskytuje sarkom z dendritických buněk, který je na rozdíl od výše uvedených dvou nemocí chemorezistentní.
Klíčová slova:
histiocytosis – histiocytóza z Langerhansových buněk – juvenilní xantogranulomatóza – Erdheimova-Chesterova choroba – sarkom z dendrických buněk
Klasifikace histiocytárních chorob
Histiocytární choroby jsou vzácné a klinickým průběhem velmi různorodé jednotky. Vycházejí ve většině případů z buněk hematopoetické řady, a proto jsou WHO klasifikací řazeny do krevních chorob. Když však jsme vzali do ruky obsáhlé učebnice hematologie, většinou jsme v nich tuto skupinu chorob nenašli.
Pro relativní vzácnost těchto chorob nejsou k dispozici spolehlivé údaje o jejich incidenci a etiopatogenetických faktorech. Jaffé v knize WHO klasifikace maligních krevních chorob udává, že tvoří 1 % ze všech maligních chorob, které vznikají v lymfatických uzlinách.
Jedinou chorobou z této skupiny, která má jasně definovaný genetický základ, je familiární erytrofagocytární lymfohistiocytóza, u níž byly prokázány mutace některých genů (gen pro perforin PRF1 a gen Munc13-4). Obě tyto mutace způsobují defektní buněčnou cytotoxicitu.
Jednotlivé formy maligních histiocytárních onemocnění je obtížné rozlišit dle klinických příznaků. Histopatologie a imunofenotypizace je proto základní diagnostickou metodou pro jejich klasifikaci.
Nemoci a syndromy, řazené do skupiny histiocytárních chorob, jsou charakteristické infiltrací a akumulací buněk monocyto-makrofágového systému v postižených tkáních. Z druhé strany, ne všechny stavy, u nichž dominuje histiocytární infiltrace, jsou klasifikovány jako histiocytóza, protože histiocytární infiltrace představuje sekundární proces. K těmto sekundárním histiocytózám patří X-vázaný lymfoproliferativní syndrom a některé vrozené lipidózy.
Pro porozumění této relativně vzácné skupině nemocí je nutné připomenout současné představy o vzniku histiocytů.
Prapůvodcem fyziologického histiocytu je kmenová buňka kostní dřeně.
Ta se vlivem určitých stimulů vyvíjí do myeloblasto-monoblastové buněčné linie. Buňky této linie se pak dále diferencují na zralé monocyty, které cirkulují v periferní krvi a následně přestupují do tkání, kde pak podstupují terminální diferenciaci. Monocyty tedy v žádném případně nerecirkulují. Po přestupu do tkání se diferencují na makrofágy kostní dřeně, Kupfferovy buňky jater, Langerhansovy buňky v kůži, mukokutánních spojích, uzlinách, plicích.
Dle funkce je možno rozdělit buňky histiocytárního systému do 2 skupin:
- antigen processing (phacocytic) cells
- antigen presenting cells neboli dendritické buňky
Dendritické buňky obvykle nefagocytují a jejich hlavní funkce je prezentovat antigen T - a B lymfocytům.
Buňky folikulárního dendritického retikula jsou v lymfatických folikulech, kde prezentují antigen B lymfoctům. Folikulární dendritické buňky neexprimují CDk45 (leukocyte common antigen), a jejich původ je proto považován spíše za stromální než hematopoetický.
Interdigitující dendritické buňky a Langerhansovy buňky jsou si v mnohém podobné a obě tyto skupiny buněk prezentují antigen T lymfocytům. Interdigitující buňky se nacházejí v parakortexu klidových a reaktivních lymfatických uzlin, Langerhansovy buňky jsou přítomné v kůži, plicích i v dalších orgánech.
Dendritické buňky nemají tolik lysozomálních enzymů jako fagocytující makrofágy. Interdigitující dendritické buňky a Langerhansovy buňky mají jen slabou lysozomální enzymovou aktivitu a folikulární dendritické buňky nemají žádnou. Všechny dendritické buňky však silně exprimují HLA antigeny první i druhé třídy.
Jednotlivé typy buněk uvádí tab. 1.

Histiocytární nemoci tvoří velmi divergentní skupinu nemocí, z nichž některé jsou reaktivní, jiné maligní a mnohdy není jasné, zda nemoc považovat za reaktivní, či maligní.
Pro popisy těchto chorob se v současnosti používá jak klasifikace, kterou vytvořila International Histiocyte Society, tak WHO klasifikace krevních chorob. Obě klasifikace uvádí tab. 2.
![Klasifikace histiocytárních chorob dle WHO klasifikace maligních krevních chorob (Jaffe, ES 2001) a dále dle International Histiocyte Society. Podle [49].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/301cd0c1cd70acd12474ce0fcd51941e.jpeg)
Histiocytóza z Langerhansových buněk (LCH)
Definice nemoci a její incidence
Uvedený termín histiocytóza z Langerhansových buněk zahrnuje široké spektrum závažnosti klinických projevů. Dříve, vzhledem ke své tajuplnosti, nejasné etiologii a rozpakům, zdali toto onemocnění řadit k nádorovým, nebo infekčním onemocněním či lipidovým tezaurismózám, bylo nazýváno histiocytosis X.
Klinicky se onemocnění manifestuje širokou škálou příznaků, počínaje náhodným RTG nálezem specifického jednoložiskového lytického procesu v kosti (označované též termínem eozinofilní kostní granulom) až po generalizované systémové postižení.
Incidence LCH v ČR zcela přesně neznáme. WHO klasifikace krevních chorob uvádí incidenci 5/1 milion všech osob s tím, že u dětí je tato nemoc podstatně častější. V České republice je dětskými hematology a onkology ročně registrováno asi 15 dětí s nově diagnostikovanou LCH, počet dětí s izolovanou kostní formou LCH, která nevyžaduje jinou než chirurgickou léčbu, není znám. Němečtí autoři uvádí incidenci LCH u dospělých 1–2 případy/1 milion obyvatel.
V dospělosti se projevuje převážně jako jedno - či víceložiskové kostní postižení, zatímco u dětí má nemoc spíše charakter systémového postižení.
Patogeneze a etiologické faktory
V případě Langerhansovy histiocytózy je stále otevřená diskuze mezi dvěma názory. Dle prvního z nich je Langerhansova histiocytóza reaktivní stav nejasného původu. Byla sice prokázána klonalita Langerhansových buněk v patologických ložiscích, ale ani tento nález zcela nevyvrátil představu reaktivního stavu a průkaz klonality není možné brát jako jednoznačný průkaz maligní etiologie. Zvažuje se také familiární predispozice, neboť určitá akumulace této poruchy v některých rodinách byla přece jenom prokázána.
Jiní autoři považují alespoň některé případy Langerhansovy histiocytózy za maligní klonální proliferaci. Na otázku, zda se jedná o maligní či reaktivní proces, není tedy zatím přijata jednoznačná odpověď. Autoři WHO klasifikace krevních chorob (2001) uvádějí definici Langerhansovy histiocytózy: „Jde o neoplastickou proliferaci Langerhansových buněk exprimujících CD1a, S-100 protein a Bierbeckova granula,“ zatímco klasifikace Mezinárodní histiocytární společnosti, event. základní učebnice dětské onkologie z roku 2006, popisují Langerhansovu histiocytózu stále jako patologický reaktivní proces neznámého původu, nikoliv jako maligní proliferaci.
WHO klasifikace krevních chorob dále definuje sarkom z Langerhansových buněk jako neoplastickou proliferaci Langerhansových buněk se zřetelnými cytologickými maligními znaky, neboli něco jako high grade histiocytózu z Langerhansových buněk.
Manifestace u dětí
Onemocnění se nejčastěji projeví ve 2–4 letech věku. U 75 % dětských pacientů postihuje histiocytóza více orgánů (kůži, kosti, uzliny, některé vnitřní orgány). Hlavní klinickou manifestací LCH u dětí je přítomnost lytických kostních lézí. Nejčastějšími prvními příznaky u kojenců a malých dětí jsou typické kožní morfy, připomínající seborrheu, typicky ve vlasaté části hlavy. Dále se mohou vyskytnout následující příznaky: chronické otitidy, příznaky diabetes insipidus, bolesti kostí, otoky hlavy, pokles víčka, event. celkové příznaky jako neprospívání, váhový úbytek, horečky, dušnost, lymfadenopatie, bolesti břicha způsobené hepatosplenomegalií, míšní kompresivní syndromy. Postižené děti obvykle málo rostou. Příčina malého růstu je asi komplexní, uvádí se nadprodukce katabolických cytokinů, vliv kortikosteroidní a cytostatické terapie a nedostatek růstového hormonu.
U malých dětí, nejčastěji mladších 2 let, je typická agresivní diseminovaná forma LCH, vyznačující se celkovým chřadnutím dítěte, hepatosplenomegalií, generalizovanou lymfadenopatií, anémií, někdy pancytopenií a případně lytickými kostními ložisky, dříve nazývaná chorobou Lettererevou-Siweho.
U starších dětí se pak popisuje formanazvaná nemoc Handova-Schüllerova--Christianova, s polyurií (diabetes insipidus z postižení pituitární oblasti), exoftalmem (způsobeným přítomností retroorbitálních granulomů) a lytickými defekty plochých kostí.
Uvedené rozdíly popsaných forem nemocí shrnuje tab. 3.

Onemocnění je léčitelné, je možné dosáhnout kompletní dlouhodobé remise či vyléčení. U dospělých, kteří měli toto onemocnění v dětství, je obvykle trvalé postižení hypotalamu a je nutná trvalá substituce antidiuretického hormonu, méně často jsou snížené hladiny i dalších hormonů.
Manifestace u dospělých
Pokud se nemoc projeví až v dospělosti, dominuje kostní manifestace. Méně častý je souběh kostního a plicního postižení. Podle analýzy provedené Baumgartnerem je postižení hypofýzy, lymfatických uzlin a jater v dospělém věku celkem vzácné, ale vyskytuje se. Při analýze projevů této nemoci u dospělých byla získána následující čísla: v 80 % bývají postiženy kosti, v 60 % kůže, ve 33 % játra, slezina a uzliny, ve 30 % kostní dřeň, ve 25 % plíce, ve 25 % orbita, ve 20 % orodentální a otologická oblast, kde může způsobit uvolňování zubů. U dospělých pacientů má onemocnění velmi různorodý průběh. U některých postihne jenom jedno ložisko a po léčbě se již neobjeví, u jiných má recidivující charakter, u pacientů jsou objevována stále nová a nová ložiska a choroba může být příčinou omezené hybnosti, či může dokonce přivodit smrt.
Diabetes insipidus bývá vyjádřen u méně než 15 % pacientů.
K prognosticky nepříznivým faktorům patří věk pod 2 roky, orgánová dysfunkce, multiorgánové postižení nebo kombinace orgánové dysfunkce s infiltrací jater.
V literatuře zatím nebylo popsáno postižení srdce, svalů, ledvin, gonád a periferních nervů. V dalším textu rozvedeme jednotlivé projevy nemoci dle orgánů a tkání, které mohou být touto nemocí postiženy.
Krev a kostní dřeň
Periferní krevní obraz je buď zcela normální, nebo je přítomna mírná anémie chronického onemocnění. Monocytóza je vzácná a spíše reaktivní. Ačkoliv prekurzory Langerhansových buněk mají svůj původ v kostní dřeni, nebyla jejich přítomnost v periferním krevním obraze zachycena. V periferním krevním obraze se udává vysoký poměr CD4/CD8 díky nízkému počtu CD8+ lymfocytů. V remisi se jejich počet vrací na normální hodnoty. Jiné typické abnormality T - a B lymfocytů nejsou patrné, koncentrace gamaglobulinů může být i zvýšená. Pancytopenie se zjistí jen u 5–10 % pacientů, u části z nich byla současně nalezena masivní infiltrace kostní dřeně Langerhansovými buňkami. U jiných naopak byla kostní dřeň normální a pancytopenie byla způsobená fagocytární aktivitou Langerhansových buněk v jednotlivých ložiscích. Stupeň postižení kostní dřeně je variabilní, u většiny pacientů Langerhansovy buňky v kostní dřeni nejsou. Masivní infiltrace kostní dřeně je výjimečná.
Kostní ložiska
Osteolytické defekty se objevují hlavně v osovém skeletu, méně v periferních kostech. Uvádí se, že kostní ložiska jsou v 51 % v oblasti kalvy, ve 30 % v čelisti (což může vést k uvolňování zubů), v 17 % v dlouhých kostech, v 13 % v obratlích, v 13 % v pánvi, 6 % v žebrech.
U některých pacientů jsou kostní ložiska různého stáří, hojící se místa mají sklerotický lem. Ne všechna musí bolet. Zduření tkání přiléhajících ke kosti signalizuje, že choroba prorůstá do okolí. Ložiska v kostech lze detekovat jak radioizotopy (technecium pyrofosfonát, FDG-PET), tak rentgenologicky. Oba typy vyšetření se doplňují. Nejcitlivější metodou zobrazení patologického děje v kosti je však magnetická rezonance (MR).
Za zvláště riziková kostní ložiska jsou považovány kostní defekty v oblasti temporální, orbitální či mastoideální. Postižení těchto kostí signalizuje vyšší pravděpodobnost pozdějšího postižení CNS, hlavně mozkového kmene a cerebella. Mnohočetnou kostní formu s postižením kalvy, pánve a femoru ilustrují obr. 1–4.


Mozek a hypofýza
Diabetes insipidus je dobře známou komplikací Langerhansovy histiocytózy, postihuje 10–50 % dětských pacientů, může se ale objevit i v dospělosti jako první příznak této nemoci. Při MR zobrazení CNS mají tito pacienti patologické infiltráty v oblasti hypotalamu.
Přibližně u 1/2 z nich se diabetes insipidus projeví jako první příznak nemoci a u dalších se objeví do 4–5 let. Postižení přední části hypofýzy je méně časté, asi u 10 % pacientů. Deficit růstového hormonu nebývá tak častý jako malý vzrůst pacientů. Příčina nedostatečného vzrůstu je asi komplexní, přispívá k němu jak léčba kortikoidy, tak nediagnostikované postižení střeva a malabsorpce. Nedostatek růstového hormonu může být důvodem neúspěšnosti in vitro fertilizace. Někdy však může vzniknout až úplný panhypopituarizmus. Hypotalamický syndrom s hyperfagií je vzácný, provází jej porucha regulace teploty a nezvyklá náladovost.
Metodou detekce těchto změn v CNS jsou jednak funkční endokrinologické testy a jednak magnetická rezonance nebo CT s aplikací kontrastní látky. Expanzivní ložiska jsou prokazatelná těmito metodami u 50 % pacientů s poruchou funkce hypofýzy. Disperzní infiltraci prokáže jedině autopsie. Není jasné, proč Langerhansovy buňky mají takovou afinitu k hypotalamu a jeho stopce.
Poškození CNS však může mít i další formy. Poměrně vzácné, ale velmi závažné, je trvalé postižení CNS. Vzhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií. Dle posledních informací je možné diferencovat 3 typy postižení:
- ohraničené granulomy v oblasti mozkové pojivové tkáně zřetelné na MR zobrazení, odpovídající tumorózním ložiskům v meningách nebo v chorioidním plexu. Jejich histologie odpovídá Langerhansovým granulomům v pojivové tkáni.
- granulomy v oblasti pojivové mozkové tkáně s částečnou infiltrací okolního nervového parenchymu jak patologickými (CD1a+), tak i reaktivními histiocyty. Tyto infiltráty byly provázené výraznou T buněčnou infiltrací a neurodegenerací, ztrátou neuronů, axonů a gliózou.
- neurodegenerativní ložiska postrádající CD1a+ buňky, nejčastěji postihující cerebellum, nucleus dentatus, cerebellární bílou hmotu a mozkovýkmen, s výraznou zánětlivou infiltrací obsahující CD8 lymfocyty. Tento pro-ces vede k degeneraci a glióze nervové tkáně. Tento neurodegenerativní proces se objevuje na základě dominantně T buněčného zánětlivého procesu a je provázena destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu.Tento typ postižení se objevuje až za dlouhou dobu od stanovení diagnózy, je postupně progredující a nevratný. Projevuje se hyporeflexií, ataxií, závratěmi, dysartrií, nystagmem, tremorem,diplopií, psychomotorickou retardací a neuropsychologickými defekty.
Uši
Poruchy sluchu mohou nastat jak postižením zevního sluchového kanálu, tak poruchou středního či vnitřního ucha propagací choroby z processus mastoideus. Infiltrace je nebolestivá a postupně vede k hluchotě. Časté jsou sekundární infekce, které jsou příčinou záměny za chronickou otitidu.
Oči
Intraokulární postižení je vzácné, zatímco infiltrace orbitálního prostoru je relativně častá. Dětští lékaři se s ní setkávají u 20–30 % nemocných Langerhansovou histiocytózou. Projevuje se ptózou víčka, edémem papily a poruchou funkce VII. nervu. Někdy může být poškozen optický nerv, což si občas kromě systémové léčby vynutí i akutní léčbu nitroložiskovou aplikací kortikosteroidů a radioterapii.
Játra a slezina
Játra i slezina mohou být touto chorobou postižena, což se projeví jejich zvětšením. Velmi silná infiltrace jater pak může vyvolat příznaky jaterního selhání (pokles albuminu, snížení aktivity koagulačních faktorů, žloutenku bez výrazného zvýšení jaterních enzymů). V chronických formách může vzniknout periportální fibrotizace s příznaky shodnými se sklerotizující cholangoitidou a obstrukční biliární žloutenkou, kterou je nutno na základně biopsie odlišit od primární sklerotizující cholangitidy a adekvátně léčit.
Plíce a dýchací cesty
Respirační cesty jsou postiženy častěji u dospělých než u dětí, 60–100 % pacientů s plicní formou jsou kuřáci. Incidence plicního postižení mezi všemi pacienty s uvedenou chorobou se udává kolem 20 %. Infiltrace vyvolává restrikční změny, které předcházejí RTG změnám. Radiografický nález je tvořen cystami a intersticiálními nodulárními opacitami obvykle blíže k hilům. Optimálním prostředkem pro diagnostiku plicní formy histiocytózy je HRCT, s jehož pomocí je možno rozpoznat tenkostěnné cysty, mikronoduly, opacity a zesílení intersticiálních prostorů. Cysty jsou přitom více koncentrované v horních lalocích, méně jich je ve středních a vynechávají kostodiafragmatický úhel. Mikronoduly miliární velikosti 0,5–4,0 mm s typickou distribucí umožní dle některých autorů rozlišit pulmonální formu LCH od jiných nodulárních chorob.
Zesílení intersticia je při HRCT vyšetření zřetelné hlavně bazálně.
Prasknutí cyst a jejich komunikace s pleurální dutinou způsobí spontánní pneumotorax. Postižení plic histocytózou často komplikuje nasedající oportunní infekce. Odlišit ji může být problém, neboť teplota a váhový úbytek mohou být jak prvními projevy plicní histiocytózy, tak mohou mít i jiné (infekční) příčiny.
V diferenciální diagnostice pak pomůže jedině bronchoalveolární laváž anebo plicní biopsie a histologické vyšetření. Samotná bronchoskopie s biopsií bronchiální sliznice je pro průkaz této nemoci zcela nepřínosná. Jedině bronchoalveolární laváž umožní identifikovat Langerhansovy buňky. Lze je identifikovat flowcytometricky průkazem buněk s pozitivitou znaku CD1a v tekutině získané bronchoalveolární laváží nebo je prokázat v sedimentu z této tekutiny elektronmikroskopickým vyšetřením a průkazem Birbeckových granulí.
Auerswald udává, že v případě plicní formy histiocytózy bývá v laváži více než 5 % CD1a+ buněk, zatímco u zdravých pacientů je počet CD1a+ menší než 1 %. Dále bývá zvýšen počet buněk v aspirované tekutině nad 106/ml s prevalencí alveolárních makrofágů. Makrofágová alveolitis je totiž přítomna u kuřáků s LCH a chybí u vzácných případů plicní LCH nekuřáků.
Dle nálezů z konce 90. let minulého století se plicní Langerhansova histiocytóza dospělých liší od ostatních forem tím, že proliferující histiocyty jsou polyklonální, zatímco u ostatních forem jsou histiocyty spíše monoklonální. Plicní forma Langerhansovy histiocytózy se považuje za reaktivní proces na kouření či jiné stimuly. U této formy bylo také potvrzeno, že v případech pacientů se silnou vůlí, kteří dokázali přestat kouřit, došlo dle RTG nálezů ke spontánní regresi nemoci.
Lymfatické uzliny
Histiocytóza obvykle nedělá výraznou adenopatii, pokud ano, jde spíše o ložiskové než generalizované postižení.
Dutina ústní a trávicí trakt
Počínající infiltrace se v této oblasti projevuje zduřením dásní a sliznice patra. Může dojít k postižení kostí a uvolňování zubů či hypertrofii dásní. Progrese infiltrátů pak vytváří ulcerace v ústech. U dětí je diagnosticky významná předčasná druhá dentice.
Sliznice střevního traktu je postižena jen zřídka. Prvními příznaky je celkové neprospívání, hubnutí. Klasické projevy malabsorpce se objevují až při rozsáhlejším postižení trávicího traktu. Anální kanál a perianální oblast jsou infiltrovány často, a tvoří tak součást kožního postižení.
Kůže
Infiltrace kůže buňkami Langerhansovy histiocytózy dominuje v oblasti mediální roviny a často postihuje intertriginózní oblasti (třísla, pupek, vulvu, perianální krajinu). Makroskopicky může být kožní postižení hodnoceno jako seborrhoický ekzém, forma kožních projevů je však pestrá. Typickou morfou pro histiocytární postižení kůže je hnědorůžová papule velikosti 1–5 mm, která může být snadno zaměněna za nespecifickou dermatitidu. U kojenců a malých dětí kožní postižení často připomíná seborrhoickou dermatitidu, typicky ve vlasaté části hlavy. Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, může však jít také o izolovanou morfu, která často spontánně regreduje. U závažných onemocnění však mohou kožní morfy splývat, ulcerovat s následnou bakteriální i mykotickou superinfekcí.
Stanovení diagnózy a rozsahu nemoci
Zásadním vyšetřením, nutným pro stanovení diagnózy, je samozřejmě bioptická excize. Pro nepatologa je vhodné jen vědět, že v době akutní choroby jsou v ložisku přítomny četné Langerhansovy buňky, exprimující proliferačními markery Ki-5l a Ki-67 a eozinofily. Později v ložisku ubývá Langerhansových buněk, přibývá makrofágů a fibrocytů a nakonec obraz odpovídá postnekrotické fibróze. Někdy může být obtížné rozlišení osteomyelitidy od kostních ložisek histiocytózy, stejně jako odlišení plicní formy histiocytózy od jiných granulomatózních plicních procesů. Histologický vývoj ložiska Langerhansovy histiocytózy lze tedy rozdělit do následujících fází:
- proliferativní stadium (převažují Langerhansovy buňky)
- granulomatózní stadium (pestrá cytologie)
- xantomatózní stadium a tvorba jizev
K potvrzení diagnózy Langerhansovy histiocytózy se proto používají další znaky, jejichž přítomnost je typická:
- protein S-100
- cytoplazmatická ATP-áza a D-manosidáza
- průkaz Birbeckových granulí v cytoplazmě elektronovou mikroskopií
- přítomnost antigenu CD1a
- přítomnost antigenu CD45
- antigen CD68 se vyskytuje nepravidelně
Diagnózu je možné ověřit také imunofenotypizací. Pro Langerhansovu histiocytózu je patognomický nález antigenů II. HLA třídy, přítomnost CD1a antigenu a často i přítomnost antigenů fyziologicky se nacházejících na fagocytujících histiocytech: CD11c a CD14. Dále jsou uváděny následující znaky CD1a++, CD11c++, CD44++ ,CD54++, CD58++. Znaky CD2, CD11a, CD11b jsou na těchto patologických buňkách výjimečně a ve slabé intenzitě.
Buňky Langerhansovy histiocytózy v kostní tkáni produkují intenzivně interleukin 1 (Il-1) a prostaglandiny, které silně stimulují osteoklasty k odbourávání kostní tkáně.
Naopak Langerhansovy buňky jsou stimulovány k proliferaci interleukinem-2, neboť pro něj mají, na rozdíl od normálních histiocytů, receptor. Další cytokiny, které in vitro a zřejmě také in vivo mohutně stimulují proliferaci těchto buněk, jsou GM CSF (Granulocyte-Macrophage Colony Stimulating Factor) a TNF α (Tumor Necrosis Factor α).
WHO klasifikace uvádí dále termín Langerhans cell sarkoma, který je vyhrazen pro maligní proliferaci Langerhansových buněk, které dle své morfologie mají známky dediferenciace a obvykle také agresivnější sarkomatózní růst a přitom imunofenotyp Langerhansových buněk.
Morfologické změny v ložisku v průběhu času, k nimž nutno přihlížet při interpretaci histologického nálezu a morfologické znaky LCH, shrnuje tab. 4.

Langerhansovu histiocytózu je možno diagnostikovat jen tenkrát, když se na ni myslí. Jinak může být zaměněna s jinými chorobami. Např. infiltrace oblasti ucha za otitis media a kožní postižení za seborrhoickou dermatitidu.
Langerhansovu histiocytózu je nutno odlišit u dětí s kožním postižením od xantogranulomu, kožní mastocytózy, různých nespecifických dermatitid a od dalších onemocnění monocyto-makrofágového systému, včetně maligních.
U nemocných s multisystémovým postižením se musí odlišit chronické myeloproliferativní onemocnění.
Vyšetření při podezření na Langerhansovu histiocytózu zahrnují: krevní obraz, jaterní enzymy, kvantitativní vyšetření bílkovin, koagulační vyšetření, snímek plic a RTG vyšetření skeletu, současně s izotopovým. Dále se při nejasnostech či patologii v krevním obraze vyšetřuje kostní dřeň, plicní funkce, osmolarita moče a případně cílené biopsie orgánů podle jednotlivých symptomů.
Pro rozsah Langerhansovy histiocytózy není vytvořen žádný všeobecně platný klasifikační systém (staging). Obecně se rozsah nemoci člení na mono a multiorgánové postižení. Tab. 5 ukazuje nové klasifikační schéma. Vlastní hodnocení stupně postižení pak nabízejí specifické léčebné protokoly, např. LCH-III, které specifikují diagnostická kritéria pro postižení jednotlivých orgánů a stratifikaci pacientů do jednotlivých rizikových skupin.
Terapie Langerhansovy histiocytózy
Lokální aplikace glukokortikoidů
Osvědčené jsou nitroložiskové injekce glukokortikoidů, např. do retrobulbárního prostoru (50–100 mg hydrokortisonu nebo 75–150 mg metylprednisolonu). Injekční ložiskovou aplikaci kortikosteroidů lze s úspěchem použít k léčbě dalších dosažitelných ložisek v měkkých tkáních či v kostech.
Cílená radioterapie
Radioterapie je vzhledem k nežádoucím účinkům v pořadí až za lokální léčbou glukokortikoidy. Ozářit je nutno ta ložiska, kam nedosáhne injekční jehla a není možná topická aplikace glukokortikosteroidů. Pokud byla provedena diagnostická operace s pravděpodobností neodstranění celé patologické tkáně, je také vhodná radioterapie.
Analýza radioterapeutických zkušeností z Německa udává při velikém rozptylu použité dávky záření medián dávky 24 Gy. Na základně analýzy použité dávky a výsledku doporučili v roce 2006 němečtí autoři pro léčbu dětí 6–10 Gy a pro léčbu dospělých 20 Gy při standardní frakcionaci.
U solitárních kostních projevů bez viscerálního postižení se běžně provádí exkochleace. Pokud není exkochleace možná, je léčbou volba solitárního ložiska radioterapie.
Substituce antidiuretického hormonu
Při diabetes insipidus nezbývá nic jiného než substituční léčba intranazální formou analogu antidiuretického hormonu, obvykle 2–3krát denně. Na místě je kompletní přešetření funkce hypofýzy, zda není deficit i ostatních hormonů.
Lokální léčení kožních forem
Kožní krusty je nutné odstranit obklady z hypermanganu anebo s přispěním dalších keratolytických prostředků. Po jejich odstranění se na ložisko histiocytózy nanáší glukokortikoidní masti nebo krémy, nezbytná je většinou i léčba superinfekcí kůže postižené LCH.
Kortikoidní kapky pomohou při postižení zevního zvukovodu.
Další možnost léčby kožních projevů histiocytózy představuje PUVA.
Systémová léčba
Pro systémovou léčbu se používají následující léky:
- glukokortikoidy
- vinka alkaloidy (vinblastin, vinkristin)
- markaptopurin etoposid
- 2-chlordeoxyadenosin
- další cytostatika
- výjimečně vysokodávkovaná chemoterapie s transplantací krvetvorných buněk
Spontánní remise se popisují u 10 až20 % onemocnění a většina dětí s LCH jinou než chirurgickou léčbu nepotřebuje, protože se u nich jedná o tzv. self-resolving proces. Proto se má s celkovou léčbou začít, až je to nezbytné. Systémová léčba se používá při viscerálním či mnoholožiskovém postižení skeletu. Nejčastěji užívanými léky, lze říci standardními, pro tuto nemoc jsou glukokortikosteroidy, vinka alkaloidy a etoposid. Jsou úspěšné u 50–60 % nemocných. Jako iniciální léčba se nejčastěji podává prednison 2 mg/kg po 4 týdny a ten se pak postupně po dalších 4–6 týdnů vysazuje. S glukokortikoidy je možno podat vinblastin (6 mg/m2 1krát týdně). Alternativou vinblastinu nebo vinkristinu je etoposid (150 mg/m2 i.v. nebo 300 mg/m2 p.o. 1., 2. a 3. den s pauzou do 21. dne). Oba režimy dosahují 60–70 % léčebných odpovědí. Po zavedení etoposidu do iniciálních léčebných schémat však nebylo dosaženo lepších výsledků, než přinesla léčba vinka alkaloidy a prednisonem. Etoposid má podstatně větší potenciál indukovat sekundární leukemie než vinblastin, a proto není začleněn do současných iniciálních schémat, používá se v rámci léčby druhé linie.
Při léčbě Langerhansovy histiocytózy byl také ověřen účinek následujících léků: cyklofosfamid, chlorambucil, cytosin arabinosid, daunorubicin, 6 merkaptopurin, metotrexát, mechlorethamin, prokarbazin. Tab. 6 uvádí léčebné postupy dle LCH-III protokolu, starší léčebné protokoly pak tab. 7.
![Přehled léčebných postupů u Langerhansovy histiocytózy dle LCH-III protokolu Podle [51]. Studie LCH-III byla zahájena 2001.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/aef1625e428534a19eb1c2a06ff5ce6c.jpeg)

Protokol první mezinárodní studie pro dospělé je z roku 2004 a vychází z výše uvedeného doporučení LCH–III.
- Postižení jednoho systému: prednison a vinblastin po 6 týdnů a následně prednison, vinblastin a merkaptorpurin po 6 měsíců.
- Multisystémová choroba: prednison a vinblastin po 6 týdnů a následně prednison, vinblastin a merkaptopurin po dobu 6–12 měsíců.
- Pacienti s pulmonální formou mají být zprvu sledováni po ukončení kouření. Pokud nemoc progreduje i po přerušení kouření, je doporučena 6měsíční léčba prednisonem.
Chemoterapeutika (vinblastin, metotrexát, cyklofosfamid, etoposid či kladribin) se doporučují až při progresi plicních ložisek navzdory ukončení kouření a prednisonové léčbě. Plicní forma LCH může totiž progredovat do terminálního stadia plicní fibrózy.
Recidiva omezená na kůži: V těchto případech byly úspěchy popisovány po perorálním metototrexátu v monoterapii či v kombinaci s merkaptopurinem nebo thalidomidem.
Interferon α
Nesplnil původní naděje v této indikaci a nyní se nepoužívá, byť v několika popsaných případech přinesla léčba INF azlepšení.
Retinoidy
Retinoid (isotretinoin, Roaccutane Roche) byl podáván u kožní formy bez kostního či viscerálního postižení. Po 8 měsících léčby isotretinoinem (1,5 mg/kg/den) kožní morfy vymizely a nemocný je po následujících 5 let bez recidivy.
2-chlorodeoxyadenosin
2-chlorodeoxyadenosin neboli cladribin je k dispozici pro pacienty s Langerhansovou histiocytózou asi od roku 1990. Cladribin má do jisté míry selektivní účinek namířený proti monocytům a lymfocytům v kterékoliv fázi buněčného cyklu. Tento účinek léku na buňky monocytární řady, kam patří i buňky Langerhansovy histiocytózy, činí z cladribinu kandidáta na lék prvé volby u všech histiocytárních chorob. V literatuře jsou popsány četné případy s výbornou léčebnou odpovědí, které dle našich zkušeností můžeme potvrdit také.
V monoterapii se používá ve stejném dávkování jako pro léčbu vlastatobuněčné leukemie. Pro léčbu LCH byla s úspěchem použita kombinace 2-chlordeoxyadenosin v klasické dávce doplněné o cytosinarabinosid 500 mg/m2 každých 12 hod po dobu 5 dní.
Thalidomid
Thalidomid má antiangiogenní a imunomodulační efekt. Je dnes jedním ze základních léků pro mnohočetný myelom. Velmi dobrý účinek byl také pozorován u kožní formy Langerhansovy histiocytózy, v krátké době 1–2 měsíců zmizely při dávkách 100–200 mg kožní morfy. U kožní formy byl popsán příznivý efekt léčby PUVA.
Transplantace kostní dřeně
Pro malou četnost Langerhansovy histiocytózy jsou i zkušenosti s vysokodávkovanou chemoterapií následovanou autologní nebo alogenní transplantací hematopoetických kmenových buněk omezené. Alogenní transpatnace představuje možnost, jak zvládnou progresivní chorobu neúspěšně léčenou konvenční léčbou. U dětí se osvědčil nemyeloablastivní přípravný režim obsahující alemtuzumab.
Bisfosfonáty
Bisfosfonáty mají svůj význam v léčbě LCH nejen proto, že mají potenciál inhibovat osteoklasty zprostředkovanou osteolýzu. Bisfosfonáty se však kumulují v retikuloendoteliálním (monocytomakrofágovém) systému, tedy i v LCH buňkách, a mají potenciál inhibovat aktivitu těchto buněk, a tedy snižovat aktivitu nemoci. Publikované zprávy hovoří o tom, že bisfosfonáty nejen mírní kostní projevy a odstraňují bolesti, ale při léčbě bisfosfonáty regredovaly i mimokostní projevy nemoci. Proto by bisfosfonáty měli dostávat všichni pacienti s kostním poškozením.
Sledování a prognóza
Monitorování aktivity Langerhansovy histiocytózy je možné pouze zobrazovacími metodami, což pro dospělého pacienta znamená zobrazení skeletu metodou scintigrafie, RTG a při nejasnostech nákladnějšími metodami, FDG-PET, CT a MR. Žádný z běžných výsledků laboratorních vyšetření nekoreluje s aktivitou nemoci a nesignalizuje remisi či recidivu. Korelace S100-β sérového proteinu s aktivitou Langerhasovy histiocytózy byla popsána poprvé v roce 2000, není však standardně prováděna.
Prognóza dětských pacientů závisí na rozsahu nemoci, ve většině případů je však choroba vyléčitelná, do dospělosti mohou přetrvávat určitá organická poškození jak chorobou, tak i léčbou. Nejdůležitějšími prognostickými faktory jsou u dětí věk při diagnóze a rozsah orgánového postižení. Mortalita je u dětí pod 2 roky významně vyšší než mortalita starších dětí, stejně tak známky postižení orgánů (zejména jater, plic a kostní dřeně) je negativním prognostickým faktorem. Prognosticky nejzávažnější se zdá být postižení jater v době diagnózy.
U dospělých je prognóza velmi divergentní, u některý osob se choroba projeví pouze jedním kostním ložiskem a již nerecidivuje, u jiných dochází k opakovaným recidivám a k závažnému poškození celistvosti skeletu.
Hemofagocytární lymfohistiocytóza
Patofyziologie hemofagocytárních lymfohistiocytóz
Hemofagocytární lymfohistiocytózapřestavuje reaktivní zmnožení lymfocytů a histocytů s probíhají hemofagocytózou.
Existuje familiární forma této nemoci s prokázanou mutací více než dvou genů, z nichž každý narušuje cytotoxickou funkci NK - a T-buněk. Defekt NK - a T-buněk má klíčovou roli pro uvedenou poruchu.
Získané formy se mohou vyskytnout u osoby s vrozenou nebo získanou poruchou imunity. Vyvolávající stimulem pak může být infekce. Podmínkou toho, aby tato získaná forma mohla vzniknout, je však výrazný defekt NK - a T-buněčné imunity.
Hereditární i získané formy se klinicky velmi podobají, a proto se pro ně používá společný termín hemofagocytární lymfohistiocytóza. Společným jmenovatelem je narušená cytotoxická funkce lymfocytů, vedoucí k přetrvávající aktivitě imunitního systému, tedy k proliferaci a akumulaci lymfocytů a histiocytů v postiženích orgánech.
Familiární hemofagocytující lymfohistiocytóza je jednotka, u níž byla prokázána mutace různých genů, které jsou důležité pro cytotoxickou funkci T - a NK-buněk.
První popsanou byla mutace genu pro perforin, další pak byla mutace genu Munc 13-4, která způsobuje defektní fúzi cytologických granulí. Následovalo odhalení dalších genů, unc13d, syntaxin 11. Také vrozené defekty imunity predisponují pro tuto nemoc (Chédiak Higashi syndrom, Griscelli syndrom 2 a na X chromozom vázaný lymfoproliferativní syndrom).
V případě získaného hemofagocytárního syndromu byla prokázána excesivní tvorba cytokinů normálními nebo maligními T lymfocyty. Kontinuálně zvýšená produkce určitých cytokinů pak indukuje hemofagocytání syndrom. Důkazem excesivní imunitní stimulace je zvýšená hladina solubilního receptoru IL 2 u pacientů s aktivní nemocí. Příznaky a jejich příčiny shrnuje tab. 8.

Charakteristickým nálezem v biopsii lymfatických uzlin je infiltrace nodulů, sinusoid, kortexu a parakortexu fagocytujícími histiocyty a zmenšení počtu lymfocytů v těchto strukturách. V patologicky zmnožených makrofázích nejsou zřetelné atypie typické pro maligní transformaci buněk. Při cytologickém hodnocení vykazují histiocyty známky aktivace, je zvýšeno množství cytoplazmy a je zřetelná fagocytóza erytrocytů, leukocytů a krevních destiček. Charakteristickým znakem je vždy smíšená lymfo-histiocytární infiltrace. Pomáhá odlišit tento typ histioctytózy od Langerhansovy histiocytózy, u níž dochází zpočátku jen ke zmnožení Langerhansových buněk.
Výrazná histiocytární proliferace je zřetelná v celém retikuloendoteliálním systému, nejvíce je postižena kostní dřeň, červená pulpa sleziny, jaterní sinusoidy a sinusoidy lymfatických uzlin. Infiltrace kostní dřeně je vždy zřetelná u hemofagocytující lymfohistiocytózy spojené s infekcí, ale může být opožděná v případě familiární formy, kdy iniciální histologie kostní dřeně může prokázat hyperplazii červené krvetvorby bez hemofagocytózy. Proto je vhodné bioptovat i jiné tkáně a orgány.
Dle souvislosti lze hemofagocytární lymfohistiocytózu dělit do 3–4 skupin:
- familiární hemofagocytární lymfohistiocytóza
- hemofagocytární lymfohistiocytóza asociovaná s infekcí
- hemofagocytární lymfohistiocytóza asociovaná s maligní neoplazií
- hemofagocytární lymfohistiocytóza asociovaná s neznámým vyvolávajícím činitelem
Familiární erytrofagocytární lymfohistiocytóza (FEL)
FEL je vzácné, často fatální multiorgánové onemocnění postihující játra, slezinu, lymfatické uzliny a centrální nervový systém. Vždy je přítomná hemofagocytóza a pozitivní rodinná anamnéza, kdy je patrný autozomálně recesivní způsob dědičnosti. Choroba se manifestuje u kojenců a batolat. Projevuje se horečkou nejasného původu, úbytkem na váze, pancytopenií a hepatosplenomegalií. Někdy lze detekovat makulopapulární exantém červeno fialového zbarvení (eflorescence u LCH bývají žluto-hnědé). FEL se obtížně diagnostikuje, neboť první bioptické vyšetření kostní dřeně a jater nezachytí hemofagocytózu. Ta je přítomna až po delším průběhu nemoci, kdy se objevuje také anémie a žloutenka. Diagnóza nemoci je přitom založena na průkazu lymfohistiocytárních infiltrátů a přítomnosti erytrocytofagocytózy ve vzorcích z lymfatických uzlin, sleziny, jater, kostní dřeně nebo plic.
Diagnostická kritéria uvádí tab. 9.
![Diagnostická kritéria fagocytární lymfohistiocytózy. Podle [52].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/e0fc7cf7503ef86bc6ce635aa5b71b90.jpeg)
Porucha se projeví náhle vzniklými horečkami a postižením výše uvedených orgánů. Možné je i postižení CNS, dezorientace, křeče, porucha vědomí, koma. Laboratorní vyšetření mohou odhalit hyperlipidemii, hypofibrinogenemii a poruchu buněčné imunity (snížení aktivity cytotoxicity). Onemocnění je autozomálně recesivně vázané. Průkaz pozitivní rodinné anamnézy může při stanovení diagnózy napomoci. Průběh nemoci je rychlý a velmi často fatální.
Klasickým lékem je etoposid, dále kortikosteroidy, vinblastin a další formy imunosuprese. Při postižení CNS se intratekálně aplikují steroidy a metotrexát. Novým a velmi účinným preparátem, podobně jako u Langerhansovy histiocytózy, je cladribin. S terapeutickým cílem byla u těchto dětí dělána také splenektomie.
Klasickým léčebným protokolem pro hemogacytující lymfohistiocytózu, který se používá jak u familiární, tak u nefamiliární formy, je trojkombinace složená z etoposidu, dexametazonu a cyklosporinu. Léčebný protokol je na www.histio.org/society/protocols se všem podrobnostmi. U méně rozvinuté dětské formy je možné použít jen kortikoidy a imunoglobuliny. Léčba druhé linie při insuficienci první není přesně definována. Formou kazuistik byla popsány případy, kdy pomohl daclizumab nebo alemtuzumab či etanercept.
Uvedená cytostatická léčba má potenciál dosáhnout u dětí zpomalení průběhu, nicméně zastavení procesu a vyléčení se uvedenou chemoterapií nepodaří vždy dosáhnout. Jediným zásadním léčebným postupem je alogenní transplantace. Ta je považována za léčbu volby, pokud je vhodný dárce.
Sekundární hemofagocytují lymfohistiocytóza
Sekundární hemofagocytující lymfohistiocytóza, asociovaná s infekcí (IAHS), byla poprvé popsána při virové infekci u imunokompromitovaného pacienta, později i u řady jiných virových, bakteriálních, mykotických a parazitárních onemocnění, provázející imunodeficitní pacienty. Podmínkou vzniku byl stav imunodeficience, a to buď vrozeného, získaného, či iatrogenního původu. IAHS lze rovněž pozorovat v souvislosti s některými T lymfoidními malignitami. Klinická symptomatologie je obdobná jako u FEL. Dominuje horečka, hepatopatie, anémie a koagulopatie. Příčina koagulopatie je zřejmě v infiltraci jater.
Základem pro stanovení diagnózy je biopsie kostní dřeně, v níž jsou benigně vyhlížející histiocytární buňky obsahující fagocytované erytrocyty a další krvinky. Fenotyp a cytochemická charakteristika je shodná s fyziologickými histiocyty a odlišná od maligních histiocytů. Podobný obraz lze nalézt i v uzlinách. V kostní dřeni může být přitom zřetelné zmnožení tvorby jak erytrocytů, tak trombocytů, přičemž v periferní krvi je jich nedostatek a nejsou přítomny specifické protilátky, které by způsobily jejich zánik na autoimunitním podkladě.
Sekundární hemofagocytární syndrom tedy není pravou maligní histiocytózou (histiocytárním sarkomem), od které musí být naopak bezpečně odlišen. Uvádíme to proto, že agresivní nástup histiocytární proliferace může vést ke zmýlení s maligní histiocytózou (generalizovaným histiocytárním sarkomem).
Léčba se zaměřuje na zvládnutí souběžně probíhající infekce, imunodeficitního stavu, případně vyvolávajícího maligního onemocnění. Pokud nemoc vznikne u pacientů na imunosupresi, je to indikací k přerušení imunosuprese.
V případech asociovaných s maligní nemocí je třeba paralelně s intenzivní symptomatickou léčbou HLH léčit i základní maligní onemocnění.
Sinusová histiocytóza s masivní lymfadenopatií (nemoc Rosei-Dorfman)
Sinusová histiocytóza s masivní lymfadenopatií byla poprvé popsána Roseiem a Dorfmanem, a tak nese jejich jméno. Tato nemoc se obvykle manifestuje nebolestivou krční lymfadenopatií, ale zvětšené mohou být uzliny i v jiných lokalizacích. Pouze u 30 % pacientů jsou postiženy mimouzlinové tkáně, kůže, kosti, prsa, může se rozšířit i na povrch hlavy a do CNS. Někdy jsou přítomna i osteolytická kostní ložiska.
Laboratorním korelátem nemoci je horečka, leukocytóza, zvýšená sedimentace erytrocytů a hypergamaglobulinemie.
V případě Rosei-Dorfmanovy nemoci nejde o smrtící chorobu a není to choroba dětí. Výskyt této nemoci kultivuje kolem 20. roku života.
Diagnózu této nemoci je možné stanovit pouze morfologicky. Pro tuto nemoc je charakteristická infiltrace uzlin v oblasti subkapsulárního sinusu fagocytárními histiocyty a dále reaktivní zánětlivé změny. Patologické histiocyty v tomto případě obsahují také protein S-100, katepsin a ojediněle dokonce antigen CD1a+, podobně jako Langerhansovy buňky, ale na rozdíl od nich neobsahují Birbeckova granula.
Z patofyziologického hlediska je zajímavé, že sinusová histiocytóza je nemoc s polyklonální proliferací patologických buněk nejasné etiologie. Původ patologických histiocytů této choroby není stanoven, nejspíše však odpovídá aktivovaným makrofágům, byť na druhé straně je zde hodně podobných znaků s Langerhansovými buňkami.
Choroba mívá u 20–50 % postižených benigní průběh se spontánní úpravou stavu. Avšak čím rozsáhlejší postižení, tím závažnější prognóza.
Vzhledem ke vzácnosti této choroby neexistují opět jednotně doporučované léčebné postupy. Úspěchy byly popsány po kortikosteroidech, alkylačních cytostatikách i po radioterapii. Z klasických léčebných postupů se proto doporučuje kombinace vinka alkaloidů, alkylačních cytostatik a glukokortikoidů. Novým lékem pro tuto nemoc je cladribin.
Juvenilní xantogranulom, nekrobiotický xantogranulom
Jde o skupinu histiocytárních chorob z fagocytujících buněk, pro něž je typická fagocytóza lipidů, což způsobuje typické zbarvení. Do této skupiny lze přiřadit: juvenilní xantogranulom a další podobná onemocnění: retikulohistiocytom, xantoma disseminatum, nekrobiotický xantogranulom či xantom. Nejčastější je juvenilní xantogranulom, který může být zaměněn výjimečně za Langerhansovu histiocytózu.
Histologickým podkladem je infiltrace podkoží pěnitými histiocyty, tvořícími xantogranulomy, jsou přítomny Teutonovy obrovské buňky, v buňkách jsou lipidové inkluze, které dávají barvu těmto ložiskům. Absence proteinu S-100 a Birbeckových granulí odlišuje tuto jednotku od Langerhansovy histiocytózy.
Uvedená onemocnění se obvykle manifestují primárně na kůži. Jejich manifestace může zůstat na kůži omezena, v těchto případech je nejčastěji postižen obličej, krajina oka, oblasti flexorů.
Kůže může být postižena pouze v malém rozsahu, ale také generalizovaně.
Ne vždy však xantogranulomatózní postižení zůstává omezeno na kůži, progresivní formy postihují nejen kůži, ale také viscerální orgány a u výjimečných forem s viscerálním postižením může kožní poškození chybět. Viscerální forma xantogranulomu se vlastně blíží Erdheimově Chesterově chorobě, která sice má primárně postihovat dlouhé kosti, ale může také infiltrovat měkké tkáně. Pokud jsou postiženy nejen kosti, ale i měkké tkáně, může mít tato nemoc fatální průběh. S narůstající mírou postižení těla se zmenšuje citlivost nemoci na léčebné zásahy.
Erdheimova-Chesterova choroba
Erdheimova-Chesterova choroba (Erdheim-Chester disease) je histiocytární onemocnění, které však nepatří k Langerhansově histiocytóze a je od ní morfologicky odlišitelné. Choroba se projevuje symetrickou sklerózou, postihující diafýzu i metafýzu dlouhých kostí, šetřící epifýzy. Radiologický nález je pro tuto nemoc patognomický. Nicméně 5–8 % pacientů může mít také postiženy ploché kosti. Erdheimova-Chesterrova choroba představuje vlastně systémovou formu xantogranulomatózního onemocnění.
Histologicky je tato nosologická jednotka definována přítomností mononukleárních infiltrátů složených z pěnitých histiocytů CD68 pozitivních, obsahujících tukové částice. Tyto buňky tvoří xantogranulomatózní a infiltrativní ložiska.
Mimokostní postižení je u této nemoci popisováno v 50 % případů. Byly popsány následující komplikace: postižení hypotalamu s následným diabetes insipidus a hypopituarizmem, retroperitonální infiltrace s postižením ledvin, případy s ložisky na očních víčkách vzhledu xantomů, exoftalmus, a také postižení plic. Plicní fibróza s dušností a srdeční selhání jsou nejčastější příčiny úmrtí. Neurologické postižení může způsobovat ataxii či parézy.
Xantogranulomatózní proces při Erdheimově Chesterově nemoci může mimo viscerální orgány či kosti postihovat také kůži a dutinu orbity.
Průběh nemoci je velmi individuální a odpovídá stupni poškození organizmu, nezřídka byl popisován fatální proces.
Nemoc se léčí podobně jako Langerhansova histiocytóza. Nicméně uvádí se, že léčebný efekt kortikosteroidů i chemoterapie je podstatně menší než u LCH. Pokud se podávaly kortikoidy, byly účinné ve vysokých pulzních dávkách. Formou case reportu byla v roce 2005 popsán úspěch interferonu au této nemoci, který navodil roky trvající remisi choroby.
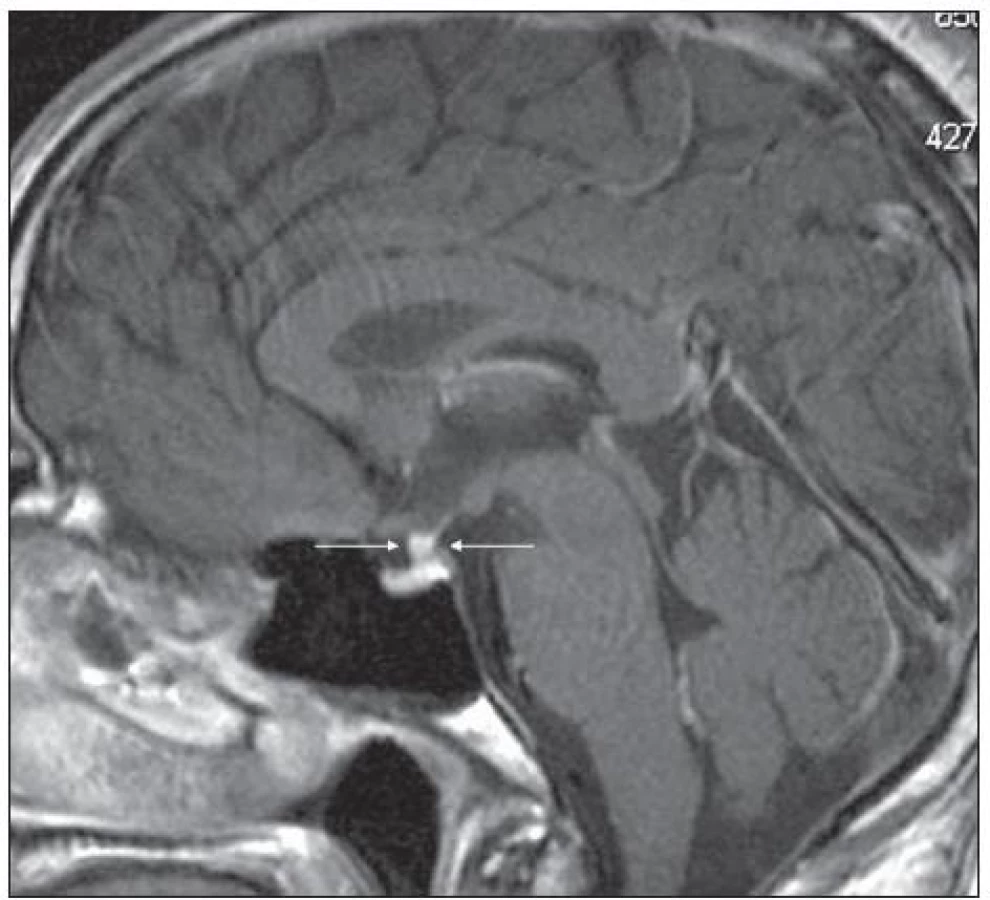
Pro Erdheimovu-Chesterovu nemoc je typická zvýšená aktivita při scintigrafii skeletu (obr. 5) a může být i infiltrovaná hypofýza s příznaky diabetu insipidus (obr. 6 a 7).


Kikuchi-Fujimoto histiocytární nekrotizující lymfadenitis
Kikuchi-Fujimoto histiocytární nekrotizující lymfadenitis je termín pro self limiting cervikální lymfadenopatii nejasného původu. Předpokládá se, že se jedná o postvirální hyperimunní reakci. Je zde možné spojení s lupus erythematosus a nespecifickými hyperimunitními reakcemi na různé vyvolávající příčiny. Klinicky se projeví jako zvětšené uzliny, nejčastěji v oblasti krku, případně spojené s horečkou nejasného původu.
Diagnózu lze stanovit pouze histologicky z extirpované uzliny. V uzlině jsou přítomna ložiska nekrózy, která mohou splývat, agretáty histiocytů a aktivované lymfocyty.
Léčba této nemoci se odvíjí od tíže příznaků. Lehčí příznaky by měla zvládnout nesteroidní antiflogistika, závažnější průběh s horečkami pak léčba glukokortikoidy. Vzhledem k tomu, že podobný obraz může mít i lupusová lymfadenitida, doporučuje se vždy vyšetření cílené na průkaz systémových chorob pojita.
Maligní histiocytární choroby
Do skupiny maligních chorob odvozených od histiocytů dle WHO patří:
- akutní monocytární leukemie
- histiocytární sarkom
- sarkom z folikulárních či interdigitujících dendritických buněk
Termín maligní histiocytóza ve WHO klasifikaci krevních chorob není. Stav, který byl dříve označován termínem maligní histiocytóza, odpovídá diseminovanému histiocytárnímu sarkomu WHO klasifikace a může se překrývat s příznaky akutní monocytární leukemie. Ta se na rozdíl od klasické myeloidní leukemie vyznačuje masivnější infiltrací nehemopoetických tkání, kůže a dásní, jater a sleziny.
Histiocytární sarkom je WHO klasifikací definován jako maligní proliferace buněk s morfologií a imunofenotypem odpovídajícím zralým tkáňovým histiocytům.
Před zavedením imunofenotypizace byly totiž četné B - i T-buněčné lymfoproliferace považovány za histiocytární malignity. Dále mnoho případů označovaných jako histiocytární medulární retikulóza a maligní histiocytóza dnes odpovídá anaplastickému velkobuněčnému lymfomu. Jiné případy histiocytární medulární retikulózy byly přehodnoceny na hemofagocytární lymfohistiocytózu.
Přibližně 1/3 histiocytárních sarkomů se manifestuje lokalizovanou lymfadenopatií, 1/3 se manifestuje kožními ložisky (solitární či mnohočetná) a poslední 1/3 vzniká extranodálně, často v oblasti zažívacího traktu.
Někteří nemocní mají systémové postižení s mnohočetnými ložisky, jehož popis se může shodovat s dřívějšími popisy maligní histiocytózy, WHO klasifikace by nyní pro tento stav použila termín generalizovaná či diseminovaná forma histiocytárního sarkomu.
Lokalizovaný maligní histiocytární sarkom
Tato jednotka je odvozena od fagocytujících mononukleárních buněk ve stadiu tkáňové fixace a diferenciace. Může vzniknout jak v kůži, tak zažívacím traktu či v kostech.
Diseminovaný maligní histiocytární sarkom, dříve zvaný maligní histiocytóza
Klinické příznaky a laboratorní nálezy
Někteří pacienti s histiocytárním sarkomem mají mnohočetné postižení včetně hepatomegalie a splenomegalie, což odpovídá staršímu popisu maligní histiocytózy. Tato diseminovaná forma histiocytárního sarkomu je velmi agresivně probíhající nemoc. Klinické příznaky se podobají projevům lymfoblastické leukemie s generalizovaným postižením orgánů. Maligní histiocytózu velmi často provází vysoké horečky nad 39 °C, splenomegalie (100 %), lymfadenopatie (92 %), hepatomegalie (67 %). Mohou však být infiltrovány i jiné orgány, např. plíce, mozek, kůže, což k výše uvedeným příznakům může přidat dušnost či bolesti hlavy. Někdy způsobuje osteolýzu a s ní spojené bolesti kostí. Kožní manifestace může být jen ve formě benigně vyhlížejícího exantému až po četné kožní tumory trupu a končetin. Postižení střeva se často projeví obstrukčními příznaky.
Nemoc charakterizují následující laboratorní změny: trombocytopenie (92 %), anémie (92 %), leukocytopenie (67 %). V biochemickém vyšetření se u těchto pacientů velmi často detekují vysoké hodnoty LDH a bilirubinu, přičemž jaterní enzymy a renální funkce bývají jen nepatrně zhoršené. Nepravidelně se vyskytuje zvýšení ACE inhibitoru (Angiotensin Converting Enzym Inhibitor) a TNF (Tumor Necrosis Factor).
Při postižení CNS lze často nalézt v mozkomíšním moku patologické fagocytující neoplastické histiocyty.
Stanovení diagnózy
Vyšetření kostní dřeně metodou trepanobiopsie je nejpřístupnější cestou ke zjištění diagnózy. Je však nutno upozornit na skutečnost, že první vzorky mohou být hodnoceny jako negativní a teprve při výraznější infiltraci se podaří identifikovat proliferující anaplastické histiocyty. Histiocytární sarkom má svou typickou morfologii a imunofenotypickou charakteristiku, popsanou ve WHO klasifikaci. Erytrofagocytóza může být přítomna, ale není dominantním jevem, zatímco je dominujícím jevem u familiární lymfohistioctózy.
Za typické histiocytární znaky je považován antigen CD68, dále lysozym, CD11c, CD14. Vzhledem k tomu, že některé jednoznačně histiocytární markery se mohou atypicky vyskytnout na myeloidních buňkách, doporučuje se při prokazování histiocytárního původu maligních buněk prokázat absenci typicky myeloidních markerů, stejně jako absenci T-buněčných a B-buněčných znaků.
Prognóza a léčba
Klinicky se tyto histocytární sarkomy chovají velmi agresivně, asi v 70 % je nemoc rozpoznána v generalizovaném stadiu (III a IV), a proto asi 60 % nemocných zemře v průběhu léčby na progresi nemoci.
Pro léčbu diseminované formy histiocytárního sarkomu (po staru maligní histiocytózy) se používají stejná cytostatická schémata jako pro léčbu agresivních lymfomů.
Také u maligní histiocytózy či histiocytárního lymfomu lze použít k léčbě cladribin.
Sarkom z folikulárních dendritických buněk
Sarkom z folikulárních dendritických buněk často (asi ve 2/3 případů) tvoří lokalizovanou lymfadenopatii, která má tendenci k lokálním recidivám po léčbě. Méně často vzniká primárně extranodálně, a to v jakékoliv lokalizaci. Tendence k diseminaci není velká.
Jaffe v knižní formě WHO klasifikace uvádí, že v 10–20 % je tento typ tumoru asociován s hyalinně vaskulárním typem Castlemanovy nemoci.
Pro sarkom z folikulárních dendritických buněk je typická pomalu rostoucí nádorová masa bez přítomnosti systémových příznaků. Sarkom z folikulárních dendritických buněk se chová indolentně, jako low grade sarkom. Většina nemocných je léčena i vyléčena kompletní chirurgickou resekcí s adjuvantní chemoterapií a radioterapií nebo bez ní. Lokální recidivy se vyskytují asi v 50 % případů a metastázy asi u 25 % případů. Asi 10–20 % nemocných s tímto typem tumoru mu po delším boji (léčbě) podlehne.
Sarkom z interdigitujících dendritických buněk
Sarkom z interdigitujících dendritických buněk je velmi vzácné onemocnění. Může vzniknout primárně v uzlině, ale i v měkkých tkáních a sami jsme se setkali s případem, kdy tumor infiltroval bérec, měkké tkáně i tibii.
Byly také popsány různé formy viscerálního postižení.
Nemoc se většinou projeví symptomatickou nádorovou masou, klasické B-symptomy jsou popisovány spíše výjimečně.
Zásadní pro léčbu je možnost provedení totální resekce. Pokud to není možné, používají se stejné chemoterapeutické režimy jako pro léčbu nehodgkinských lymfomů. Uvádí se, že efekt samotné chemoterapie není tak dobrý, jako je u maligních lymfomů. Transplantace kostní dřeně je proto vždy ke zvážení, pokud není možná radiální operace a odstranění patologické masy.
Prognóza této nemoci je nejistá.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e mail: z.adam@fnbrno.cz
Doručeno do redakce: 25. 2. 2009
Zdroje
1. Adam Z, Křivanová A. Léčba recidivující formy multifokální histiocytózy z Langerhansových buněk 2-chlordeoxyadenosinem u osob dospělého věku. Zkušenosti jednoho pracoviště. Transfuze Hematol 2007; 13 : 63–66.
2. Adam Z, Kavan P, Koutecký J et al. Histiocytóza z Langerhansových buněk. Čs Pediat 1997; 52 : 899–905.
3. Adam Z, Kavan P, Vorlíček J et al. Histiocytární choroby. Klin Onkol 1997; 10 : 167–173.
4. Adam Z, Vaníček J, Šlampa P et al. Histiocytóza z Langerhansových buněk u osob dospělého věku – zkušenosti jednoho pracoviště. Vnitř Lék 2006; 52 : 355–370.
5. Allen CE, McClain KL. Langerhans cell histiocytosis: a review of past, current and future therapies. Drugs Today (Barc) 2007; 43 : 627–643.
6. Allen TC. Pulmonary Langerhans cell histiocytosis and other pulmonary histiocytic diseases: a review. Arch Pathol Lab Med 2008; 132 : 1171–1181.
7. Anton M, Holoušova M, Řehůřek J et al. Histiocytoza X a dětská očnice. Čs Ophthal 1992; 48 : 176–180.
8. Arceci RJ, Brenner MK, Pritchard J et al. Contraversis and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 339–357.
9. Arceci RJ. When T cells and macrophages do not talk: the hemophagocytic syndromes. Curr Opin Hematol 2008; 15 : 359–367.
10. Bittenglova R, Pešek M, Mukenšnabl P et al. Granulomatóza z Langerhansových buněk. Stud pneumol phtiseol 2002; 62 : 196–202.
11. Bunanska E, Stančokova T, Dluholucky S. Histiocytóza z Langerhansových buněk. Čes slov Pediat 1998; 53 : 18–19.
12. Cao D, Ma J, Yang X et al. Solitary juvenile xanthogranuloma in the upper cervical spine: case report and review of the literatures. Eur Spine J 2008; 17 (Suppl 2): S318–S323.
13. Cooper N, Rao K, Goulden N et al. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant 2008; 42 (Suppl 2): S47–S50.
14. Cosgrove GP, Frankel SK, Brown KK. Challenges in pulmonary fibrosis. 3: Cystic lung disease. Thorax 2007; 62 : 820–829.
15. Costabel U, Guzman J, Bonella F et al. Bronchoalveolar lavage in other interstitial lung diseases. Semin Respir Crit Care Med 2007; 28 : 514–524.
16. Créput C, Galicier L, Buyse S et al. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med 2008; 34 : 1177–1187.
17. De Pas T, Spitaleri G, Pruneri G et al. Dendritic cell sarcoma: an analytic overview of the literature and presentation of original five cases. Crit Rev Oncol Hematol 2008; 65 : 1–7.
18. Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am 2008; 28 : 293–313.
19. Gasent Blesa JM, Alberola Candel V, Solano Vercet C et al. Langerhans cell histiocytosis. Clin Transl Oncol 2008; 10 : 688–696.
20. Gupta P, Babyn P. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a clinicoradiological profile of three cases including two with skeletal disease. Pediatr Radiol 2008; 38 : 721–728.
21. Haroche J, Amoura Z, Touraine P et al. Bilateral adrenal infiltration in Erdheim--Chester disease. Report of seven cases and literature review. J Clin Endocrinol Metab 2007; 92 : 2007–2012.
22. Hattori T, Takahashi A, Nagai Y et al. Subcutaneous juvenile xanthogranuloma. Eur J Dermatol 2008; 18 : 189–190.
23. Janka GE. Hemophagocytic syndromes. Blood Rev 2007; 21 : 245–253.
24. Kairouz S, Hashash J, Kabbara W et al. Dendritic cell neoplasms: an overview. Am J Hematol 2007; 82 : 924–928.
25. La Barge DV 3rd, Salzman KL, Harnsberger HR et al. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): imaging manifestations in the head and neck. AJR Am J Roentgenol 2008; 191: W299–W306.
26. Lin YW, Horiuchi H, Ueda I et al. Recurrent hemophagocytic lymphohistiocytosis accompanied by Kikuchi’s disease. Leuk Lymphoma 2007; 48 : 2447–2451.
27. Luz FB, Kurizky PS, Ramos-e-Silva M. Reticulohistiocytosis. Dermatol Clin 2007; 25 : 625–632.
28. Wang CW, Colby TV. Histiocytic lesions and proliferations in the lung. Semin Diagn Pathol 2007; 24 : 162–182.
29. Makras P, Alexandraki KI, Chrousos GP et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 2007; 18 : 252–257.
30. Matsui K, Nagata Y, Hiraoka M. Radiotherapy for Erdheim-Chester disease. Int J Clin Oncol 2007; 12 : 238–241.
31. Mottl H, Koutecký J, Ganevová M. Strategie léčby histiocytózy z Langerhansových buněk u dětí. Čsv Pediat 1994; 49 : 81.
32. Mottl H, Mracek J, Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530–533.
33. Mottl H, Starý J. Histiocytóza z Langerhansových buněk u dětí – klinická diagnostika a současná léčba. Čes slov Pediatr 2007; 62 : 200–225.
34. Pacovska V, Bortlova A, Homolka J et al. Granulomatóza z Langerhansových buněk. Trendy Med 2002; 4 : 59–61.
35. Rao RN, Goodman LR, Tomashefski JF Jr. Smoking‑related interstitial lung disease. Ann Diagn Pathol 2008; 12 : 445–457.
36. Rožánek P, Molnar V, Rešl M. Tři případy plicní granulomatózy z Langerhansových buněk. Lék Zpr 1998; 43 : 127–132.
37. Rupp D, Molitch M. Pituitary stalk lesions. Curr Opin Endocrinol Diabetes Obes 2008; 15 : 339–345.
38. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol 2008; 25 : 291–295.
39. Shimada A, Kato M, Tamura K et al. Hemophagocytic lymphohistiocytosis associated with uncontrolled inflammatory cytokinemia and chemokinemia was caused by systemic anaplastic large cell lymphoma: a case report and review of the literature. J Pediatr Hematol Oncol 2008; 30 : 785–787.
40. Skácel Z, Marel M, Vraštilová P et al. Histiocytóza z Langerhansových buněk: přehled literatury a vlastní pozorování. Stud pneumol phtiseol 2000; 60 : 150–156.
41. Smilek P, Krejčova B, Čada K et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994; 43 : 263–265.
42. Stover DG, Alapati S, Regueira O et al. Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer 2008; 51 : 130–133.
43. Šímová B, Mališ J, Neuwirt J. Klinické projevy histiocytózy z Langerhansových buněk. Lék Listy 2003; 49 : 10.
44. Tichy M jr, Tichy M, Krč I et al. Multicentrická retikulohistiocytóza. Cs Derm 1999; 74 : 168–171.
45. Toušovská K, Slavík Z. Histiocytóza z Langerhansových buněk v dětském věku. Lék Zprav 1997; 42 : 127–132.
46. Vaníček J, Krupa P, Adam Z. Diagnostika postižení kostí maligní chorobou. Postgrad Med 2006; 8 : 259–264.
47. Večer J, Charvát J, Kubatova H et al. Sekundární hemofagocytující syndrom při systémovém onemocnění. Čas Lék čes 2000; 139 : 379–381.
48. Weitzman S, Egeler RM. Langerhans cell histiocytosis: update for the pediatrician. Curr Opin Pediatr 2008; 20 : 23–29.
49. Favara EB et al. Contemporary classification of histiocytis disorders. Med Pediatr Oncol 1997; 29 : 157–166.
50. Adult Langergans cell histiocytosis. Eur J Haematol 2006; 76 : 363–368.
51. McClain KL. Drug therapy for the treatment of Langerhans cell histiocytosis. Expert Opin Pharmacother 2005; 6 : 2435–244.
52. Henter et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2006.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2009 Číslo Supplementum 1
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Diferenciální diagnostika eozinofilie
- Přetížení železem – novinky v patogenezi a léčbě
- Histiocytární choroby
- Akutní krvácení z horní části gastro intestinálního traktu