
Mikrodeleční syndrom Xp21: Závažná příčina selhání nadledvin, svalové dystrofie, poruchy hladin krevních lipidů a vývojové retardace u dvouměsíčního neprospívajícího kojence
Xp21 microdeletion syndrome: Severe cause of adrenal insufficiency, muscular dystrophy, plasma lipid disorder and developmental delay in a two-month-old child with failure to thrive
We report a male patient presenting at the age of 2 months with failure to thrive, vomiting, dystrophy and hypotonia. The infant with unapparent family history was delivered 2 weeks post term after an uneventful pregnancy without perinatal complications. The first laboratory investigation showed severe hyponatremia (120.0 mmol/l) and hyperkalemia (7.0 mmol/l), prompting the working diagnosis of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency and leading to intravenous supplementation of NaCl. However, subsequent investigations repeatedly revealed very low levels of plasma 17alpha-hydroxyprogesterone, making the 21-hydroxylase deficient form of CAH unlikely. Additionally, our patient had elevated muscle enzymes (CK 37.72 µkat/l) and myoglobin (212 µg/l), as well as hypertriglyceridemia (8.67 mmol/l) and high levels of glycerol in plasma and urine. At this point, the diagnosis of Xp21 contiguous gene deletion syndrome was considered, which is recognized as a microdeletion syndrome involving contiguous loci on the short arm of the X chromosome, including the genes DAX1 for congenital adrenal hypoplasia (AHC), DMD (Duchenne muscular dystrophy) and GK (glycerol kinase deficiency). Owing to this suspicion, ACTH test was performed with abnormal results (serum cortisol 125 mmol/l at 60th minute after application of Synacthen®), confirming the adrenal insufficiecy and leading to the initiation of therapy with mineralo - and glucocorticoids on which the ionic dysbalance improved.
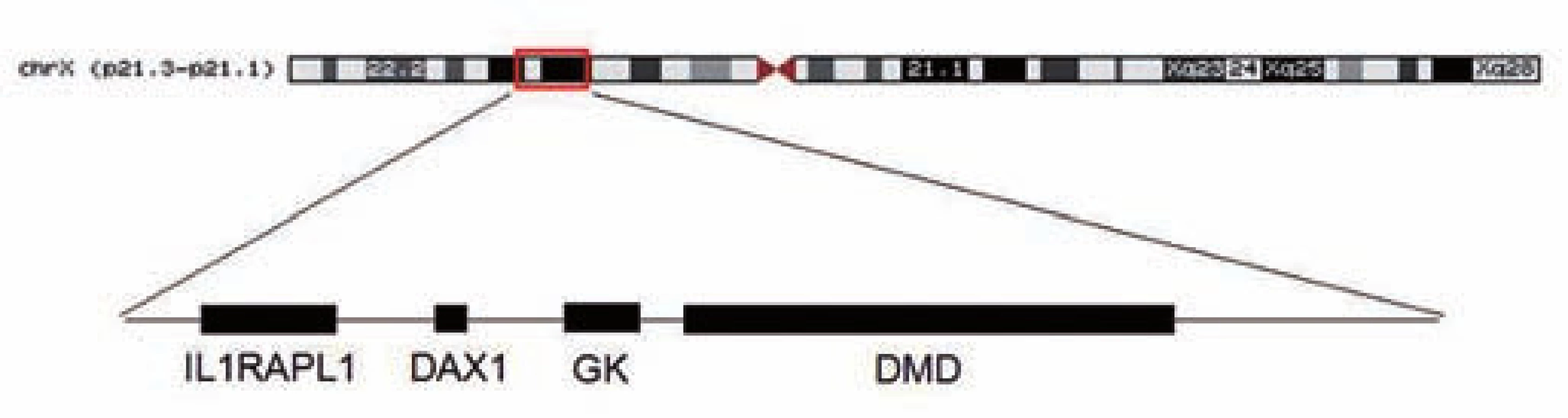
Finally, a genetic study using MLPA, PCR and FISH method revealed a deletion of 8.7 Mb including DMD, DAX1, GK and IL1RAPL1 gene (associated with X-linked mental retardation). The karyotype of our patient is 46,XY,del(X)(p21.2p21.3).
The patient was on long-term substitution with gluco - and mineralocorticoids, had normal serum electrolytes and performed rehabilitation. His long-term prognosis was unfavourable due to a complete DMD gene deletion. He suddenly died probably due to a cardiac arrest within an adrenal crisis during an intercurrent infection at 3 years of age.
Key words:
microdeletion syndrome Xp21, congenital adrenal hypoplasia, Duchenne muscular dystrophy, glycerol kinase deficiency
Autori:
F. Fencl 1; R. Průša 2; K. Banghová 1; K. Bláhová 1; Š. Vejvalková 3; S. Koloušková 1; J. Lebl 1
Pôsobisko autorov:
Pediatrická klinika UK 2. LF a FN Motol, Praha, přednosta prof. MUDr. J. Lebl, CSc., Ústav klinické biochemie a patobiochemie UK 2. LF a FN Motol, Praha , přednosta prof. MUDr. R. Průša, CSc., Ústav biologie a lékařské genetiky UK 2. LF a FN Motol,
1
Vyšlo v časopise:
Čes-slov Pediat 2012; 67 (1): 33-37.
Kategória:
Kazuistika
Súhrn
Dvouměsíční chlapec s nevýznamnou rodinnou a perinatální anamnézou byl vyšetřován pro neprospívání, dystrofii, zvracení a hypotonii. Při přijetí k hospitalizaci byla laboratorně zjištěna těžká hyponatrémie (120,0 mmol/l) a hyperkalémie (7,0 mmol/l). Bezprostředně byla zahájena parenterální korekce iontogramu a vyloučena kongenitální adrenální hyperplazie (CAH) způsobená deficitem 21-hydroxylázy (opakovaně nízké hodnoty 17alfa-hydroxyprogesteronu). Při dalším vyšetřování byly zjištěny vysoké hodnoty svalových enzymů (CK 37,72 µkat/l, CK-MB 99 µg/l), myoglobinu (212 µg/l), izolovaná hypertriacylglycerolémie (8,67 mmol/l) a vysoké hodnoty glycerolu v plazmě a moči. Na základě této kombinace příznaků bylo vysloveno podezření na diagnózu mikrodelečního syndromu Xp21 (Xp21 contiguous gene deletion syndrome), který postihuje sousedící geny DAX1 (zodpovědný za kongenitální adrenální hypoplazii – AHC), DMD (Duchennova svalová dystrofie) a GK (deficit glycerolkinázy) na krátkém raménku X chromozomu. Patologický výsledek ACTH testu (sérový kortizol 125 mmol/l za 60 minut po podání 0,75 mg Synacthenu®) potvrdil adrenální insuficienci. Byla zahájena substituční terapie mineralokortikoidy a glukokortikoidy, která vedla k úpravě iontogramu. Genetické vyšetření za použití metod MLPA, PCR a FISH následně prokázalo deleci 8,7 Mb úseku, který zahrnoval geny DMD, DAX1, GK a IL1RAPL1 (asociován s X-vázanou mentální retardací). Karyotyp chlapce byl tedy 46,XY,del(X)(p21.2p21.3).
Pacient byl dále léčen substitučními dávkami gluko - a mineralokortikoidů, při kterých byl iontogram v normě. Hmotnostně prospíval a rehabilitoval, ale jeho životní prognóza nebyla příznivá vzhledem k úplné deleci genu DMD. Ve věku 3 let došlo k náhlému úmrtí, pravděpodobně v důsledku srdeční zástavy při adrenální krizi během interkurentního infektu.
Klíčová slova:
mikrodeleční syndrom Xp21, kongenitální adrenální hypoplazie, Duchennova svalová dystrofie, deficit glycerolkinázy
ÚVOD
Xp21 mikrodeleční syndrom (Xp21 countiguous gene syndrome, MIM ID #300679) je X-vázané recesivní onemocnění, jeho symptomatologie vyplývá z rozsahu mikrodelece. Oblast p-raménka X chromozomu obsahuje v blízkém sousedství řadu genových lokusů zahrnujících zejména geny DAX1, DMD, GK [1, 2]. Gen DAX1 (dosage-sensitive sex-reversal adrenal hypoplasia critical region on the X chromosome protein 1) kóduje protein ze skupiny jaderných receptorů, který funguje jako transkripční supresor [3]. Je exprimován v řadě orgánů – kůře nadledvin a gonádách (společně původem z urogenitální lišty), hypotalamu a adenohypofýze. Jeho defekt v důsledku mutace nebo delece vede k poruše vývoje nadledvin a ke kongenitální adrenální insuficienci (AHC, adrenal hypoplasia congenita) v důsledku chybění permanentní dospělé kortikální zóny [4]. AHC se obvykle manifestuje v časném kojeneckém věku adrenální krizí s minerálním rozvratem. Později tento genový defekt vede k hypogonadotropnímu hypogonadismu a primární poruše spermatogeneze [5]. Gen DMD (Duchenne muscular dystrophy) kóduje protein dystrofin, důležitou strukturální komponentu cytoskeletu buněk příčně pruhovaného svalu. Jeho deficit je zodpovědný za vznik X-vázaných degenerativních svalových onemocnění, Duchennovy, resp. Beckerovy muskulární dystrofie. Incidence je udávána 1 : 3500 chlapců [6]. Produktem genu GK je protein glycerolkináza, jejíž izolovaný deficit vede k pseudohypertriacylglycerolémii, hyperglycerolémii a hyperglycerolurii, u novorozenců s izolovaným deficitem glycerolkinázy byly občas popsány hypotonie a apnoické pauzy. Izolovaný deficit glycerolkinázy nevede k psychomotorické retardaci ani klinickým projevům [7].
AHC, Duchennova svalová dystrofie a deficit glycerolkinázy mohou být izolovanými onemocněními při deleci nebo mutaci postihující pouze jeden gen, mohou však být součástí Xp21 mikrodelečního syndromu, kdy delece postihuje více sousedních genových lokusů. Klinický obraz potom odpovídá počtu postižených genů [8, 9]. Typicky bývá součástí mikrodelece i oblast chromozomu spojená s X-vázanou mentální retardací.
KAZUISTIKA
Pacient se narodil ze třetí fyziologické gravidity, v termínu, zdravým nepříbuzným rodičům. Jedno z předchozích těhotenství bylo pro Downův syndrom uměle ukončeno, z dalšího těhotenství se narodil o čtyři roky starší zdravý chlapec. Porodní míry pacienta byly 3520 g a 49 cm, poporodní adaptace proběhla bez komplikací. Z porodnice byl propuštěn s hmotností 3200 g jako fyziologický novorozenec.
Ve věku 2 měsíců byl přijat do okresní nemocnice jako doprovod nemocné matky. Při přijetí byl nápadně bledý, dystrofický (hmotnost 3190 g), opakovaně zvracel a měl svalovou hypotonii. V laboratorním vyšetření dominovala těžká hyponatrémie 120,0 mmol/l, hyperkalémie 7,0 mmol/l, vysoké hladiny aminotransferáz (AST 3,72 μkat/l, ALT 4,42 μkat/l) a zvýšená urea (9,4 mmol/l). Bezprostředně po překladu na naše pracoviště (obr. 1, 2) byla zahájena parenterální korekce iontogramu a vyloučena kongenitální adrenální hyperplazie (CAH) způsobená deficitem 21-hydroxylázy – hladiny 17alfa-hydroxyprogesteronu byly opakovaně nízké. Další biochemická vyšetření ukázala svalový původ transamináz a vysoké hodnoty dalších svalových enzymů (CK 37,72 μkat/l, CK-MB 99 μg/l), myoglobinu (212 μg/l), rovněž izolovanou hypertriacylglycerolémii (8,67 mmol/l) a vysoké hodnoty glycerolu v plazmě a moči (hladina v moči 102 mmol/l). Sérum bylo čiré, v elektroforéze lipoproteinů byl normální nález. Endokrinologická vyšetření potvrdila významně zvýšenou hladinu ACTH (292 pg/ml) a plazmatické reninové aktivity (51,6 μg/l/h). Na základě této kombinace příznaků bylo vysloveno podezření na mikrodeleční syndrom Xp21 (Xp21 contiguous gene deletion syndrome), který postihuje sousedící geny DAX1 (zodpovědný za kongenitální adrenální hypoplazii – AHC), DMD (Duchennova svalová dystrofie) a GK (deficit glycerolkinázy) na krátkém raménku X chromozomu. Vzhledem k podezření na AHC byl proveden ACTH stimulační test s podáním Synacthenu®, jehož patologický výsledek AHC jednoznačně potvrdil (sérový kortizol 150 mmol/l na počátku testu a 125 mmol/l za 60 minut po podání 0,75 mg Synacthenu®). Bezprostředně po tomto testu byla proto zahájena substituční terapie mineralokortikoidy (Fludrocortison v dávce 100 μg/den) a glukokortikoidy (Hydrocortison v dávce 5 mg/den), která vedla spolu s perorální substitucí natria k úpravě iontogramu.


Genetické vyšetření za použití metod MLPA (multiplex ligation-dependent probe amplification), PCR (polymerase chain reaction) a FISH (fluorescence in situ hybridization) potvrdilo očekávanou deleci 8,7 Mb úseku, který zahrnoval geny DMD, DAX1, GK a IL1RAPL1 (asociován s X-vázanou mentální retardací). HRT (high resolution technique) potvrdila deleci X chromozomu, karyotyp chlapce byl tedy 46,XY,del(X)(p21.2p21.3) (obr. 3). U matky pacienta bylo potvrzeno nosičství této mikrodelece.
Po propuštění z hospitalizace pacient zůstal v ambulantním endokrinologickém sledování, jeho iontogram byl v normě, váhově začal dobře prospívat a rehabilitoval podle Vojty. S postupujícím věkem začala být nápadná psychomotorická retardace – ve 4 měsících ještě nezvedal hlavičku, v 10 měsících se přetáčel ze zad na břicho, v 15 měsících začínal lézt pomocí rukou, ve 2 letech vývoj odpovídal 4. trimenonu, dokázal se sám posadit, sed pevný, u opory naznačuje vzpřimování, objevuje se strabismus (obr. 4), ve 3 letech si sám stoupal, chůze při držení za obě ruce, objevuje se pseudohypertrofie lýtek.

Dlouhodobá prognóza byla nepříznivá zejména vlivem úplné delece genu DMD, která vede k nejtěžší formě Duchennovy svalové dystrofie a předčasnému úmrtí. Ve věku 3 let chlapec náhle zemřel, pravděpodobně na srdeční zástavu při hyperkalémii vlivem iontového rozvratu během interkurentního infektu.
DISKUSE
V této kazuistice demonstrujeme velmi vzácný případ kombinace AHC, Duchennovy muskulární dystrofie a defektu glycerolkinázy na podkladě Xp21 mikrodelečního syndromu u dvouměsíčního kojence. Dominujícím klinickým symptomem bylo neprospívání, dystrofie a zvracení. Vstupní laboratorní nálezy potvrdily adrenální krizi s minerálním rozvratem. U chlapců s mutací nebo delecí DAX1 genu, která způsobuje AHC, bývá solná krize přítomna až v 60 % v průběhu prvních 2 měsíců života [10]. Méně obvyklá bývá manifestace onemocnění v dětství, vzácně k ní může dojít až v dospělosti [11]. Literárně popisovaná hypoglykémie nebyla u našeho pacienta zachycena.
Stanovená vysoká hodnota triacylglycerolů (TG) 8,67 mmol/l u našeho pacienta byla falešným nálezem, ve skutečnosti se při použití dané laboratorní metodiky stanovuje glycerol. Jedná se tedy o tzv. pseudohypertriacylglycerolémii, která je typická pro osoby s deficitem glycerolkinázy. Koncentrace triacylglycerolů je obvykle stanovována po odštěpení mastných kyselin lipázou enzymovou fotometrickou metodou (s glycerol-3-fosfát oxidázou). Metoda je zatížena vždy systematickou pozitivní chybou způsobenou různým obsahem volného glycerolu v séru každého jedince. Tato chyba většinou nepřesahuje 0,1 mmol/l a pro rozhodování o diagnostice a léčbě ji lze většinou zanedbat. Při deficienci glycerolkinázy dosahují triacylglyceroly vysokých hodnot, jedná se o pseudohypertriacylglycerolémii, která, pokud není správně diagnostikována, může vést k chybné léčbě hypolipidemiky (fibráty). V dospělosti se jedná o relativně častý náhodný nález. Tuto diagnózu potvrdí čiré sérum a normální elektroforetický nález lipoproteinů. Hyperglycerolémii a hyperglycerolurii u našeho pacienta následně potvrdilo podrobnější metabolické vyšetření.
Zatímco AHC je nejrizikovější komponentou Xp21 mikrodelečního syndromu v kojeneckém období (solná krize) a později možností minerálního rozvratu v obdobích interkurentních infektů, z hlediska dlouhodobého přežívání je limitující Duchennova muskulární dystrofie. Vzhledem k úplné deleci DMD genu se rozvíjí nejtěžší forma svalového postižení, která vede k úmrtí nejpozději na počátku 3. dekády života.
Z klinického hlediska je velmi významný průkaz nosičství mikrodelece Xp21 u matky pacienta, zejména v souvislosti s možností genetického poradenství a prenatální diagnostiky v případě následné gravidity.
K úmrtí pacienta došlo náhle v domácím prostředí v průběhu virového respiračního infektu. Sekční nález byl ve shodě se základní diagnózou Duchennovy svalové dystrofie, ale bezprostřední příčinu smrti neobjasnil. Nejpravděpodobnější je srdeční zástava při hyperkalémii, ke které mohlo dojít v rámci minerálního rozvratu při febrilním stavu při relativně nedostatečné dávce kortikoidů.
ZÁVĚR
Xp21 mikrodeleční syndrom zahrnující AHC může být jednou z příčin minerálního rozvratu u chlapců v kojeneckém věku. I když je velmi vzácný, je nutné o něm v diferenciální diagnostice u postižených chlapců uvažovat. Jednoznačnou diagnostickou metodou pro průkaz AHC je stimulační synacthenový test. Pseudohypertriacylgylcerolémie je naopak relativně častějším nálezem, který nevyžaduje další terapii.
Podpořeno granty MSM 0021620814, CHERISH a MZOFNM2005.
Došlo: 16. 5. 2011
Přijato: 29. 7. 2011
MUDr. Filip Fencl, Ph.D.
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: filip.fencl@gmail.com
filip.fencl@lfmotol.cuni.cz
Zdroje
1. Sjarif DR, Ploos van Amstel JK, Duran M, Beemer FA, Poll-The BT. Isolated and contiguous glycerol kinase gene disorders: a review. J Inherit Metab Dis 2000; 23(6): 529–547.
2. Stanczak CM, Chen Z, Zhang YH, Nelson SF, McCabe ER. Deletion mapping in Xp21 for patients with complex glycerol kinase deficiency using SNP mapping arrays. Hum Mutat 2007; 28(3): 235–242.
3. Skinningsrud B, Husebye ES, Gilfillan GD, Frengen E, Erichsen A, Gervin K, et al. X-linked congenital adrenal hypoplasia with hypogonadotropic hypogonadism caused by an inversion disrupting a conserved noncoding element upstream of the NR0B1 (DAX1) gene. J Clin Endocrinol Metab 2009; 94(10): 4086–4093.
4. Wheeler B, George PM, Mackenzie K, Hunt P, Potter HC, Florkowski CM. Three cases of congenital adrenal hypoplasia with novel mutations in the (NROB1) DAX-1 gene. Ann Clin Biochem 2008; 45(Pt 6): 606–609.
5. Lebl J, Jufieda K, Kalvachová B, Votava F. Chlapec s adrenální insuficiencí a defektem DAX-1 genu. Diabetologie, Metabolismus, Endokrinologie, Výživa 2001; 2 : 136–139.
6. Seemanová E, Hedvičáková P. Duchennova muskulární dystrofie u dívky. Čes-slov Pediat 2007; 4 : 234–238.
7. Lewis B, Harbord M, Keenan R, Carey W, Harrison R, Robertson E. Isolated glycerol kinase deficiency in a neonate. J Child Neurol 1994; 9(1): 70–73.
8. Darras BT, Francke U. Myopathy in complex glycerol kinase deficiency patients is due to 3’ deletions of the dystrophin gene. Am J Hum Genet 1988; 43(2): 126–130.
9. Seltzer WK, Angelini C, Dhariwal G, Ringel SP, McCabe ER. Muscle glycerol kinase in Duchenne dystrophy and glycerol kinase deficiency. Muscle Nerve 1989; 12(4): 307–313.
10. Ozer EA, Kaya A, Yildirimer M, Guler O, Can S, Aydinlioglu H. A novel DAX1 gene mutation in a Turkish infant with X-linked adrenal hypoplasia congenita. Eur J Pediatr 2009; 168(3): 367–369.
11. Yang F, Hanaki K, Kinoshita T, Kawashima Y, Nagaishi J, Kanzaki S. Late-onset adrenal hypoplasia congenita caused by a novel mutation of the DAX-1 gene. Eur J Pediatr 2009; 168(3): 329–331.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2012 Číslo 1
- Léčba bolesti a horečky u dětí
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- Očkování nejvíc potřebuje ten, kdo sám být očkován nemůže − kazuistika
- Pokrok v boji s malárií − první vakcína poskytující přijatelnou ochranu proti nemoci
Najčítanejšie v tomto čísle
- Parenterální výživa
- Kdy vyšetřovat vrozená trombofilní rizika u dětí?
- Ovlivnění bolesti při stomatologickém ošetření dětí
- Cmúľanie prstov a jeho vplyv na vývoj tváre a dutiny ústnej v detskom veku