
Klinicky relevantné možnosti a limity diferenciálnej diagnostiky megaloblastovej anémie a myelodysplastického syndrómu typu refraktérnej anémie v trepanobioptických vzorkách kostnej drene
Clinically relevant possibilities and limits of differential diagnosis of megaloblastic anemia and myelodysplastic syndrome – refractory anemia type in bone marrow biopsies
Introduction:
Megaloblastic anemia (MA) represents a subtype of macrocytic anemia caused by impaired DNA synthesis, mostly due to folate and vitamin B12 deficiency. Its mildest forms lead to macrocytosis without concomitant anemia, but more severe forms to thrombocytopenia and/or leucopenia as well. In majority of the cases, the diagnosis of MA dose not represent a serious clinical problem, however, other causes of macrocytosis including myelodysplastic syndrome (MDS) must be excluded.
Material and methods:
In the period 2004–2015 we identified in our registry 126 consecutive bone marrow (BM) biopsies of patients with cytopenia/s in peripheral blood and suspicion either on MA or MDS of refractory anemia (RA) type. We performed a retrospective analysis of BM biopsies focused on evaluation of parameters useful for the differential diagnosis, as represented by (a) cellularity and proportions of BM precursors, (b) and their topography, (c) presence of maturation defects and dysplastic changes, (d) grade and extent of myelofibrosis, (e) iron deposits and (f) presence of “inflammatory” response in BM. Histological analyses were supported by immunohistochemical examinations.
Results:
The series consisted of biopsies of 126 patients (61 men and 65 women) with average age 63 (14–88 years) – almost all patients (121/126) presented with anemia. Based on the findings we distinguished three diagnostic groups – MA (31 patients), MDS-RA (39) and bioptically unclasifiable case (“DIF DG” – 56 patients). Abnormalities of the BM cellularity were observed in 81 % and of topography in 73 % of all cases respectively. Megalobastic differentiation of erythropoesis was detected in 79 % and diagnostic dysplastic changes in 25 % of all biopsy cases. In 29 % of all biopsies ring sideroblasts were present, megakaryocytic nuclear lobulisation defects density changes were found in 61 % of all patients. In 14 % of all biopsies the BM myelofibrosis was absent, in contrast 5 % of the biopsies showed severe diffuse fibrosis. “Inflammatory” response was developed in 44 % of all biopsies. Iron deposits were absent in 26 %, decreased in 35 % and increased in 33 % of all the cases.
Conclusions:
From the point of view of histopathologist it seems to be difficult to distinguish BM hematopoietic changes in patients with MA and MDS-RA respectivelly, as histological examinations allowed determination of a definitive and correct diagnosis in about 55% of the cases. The crucial problem represents a decision whether the observed changes really result from the development of a clonal disease.
Key words:
megaloblastic anemia – megaloblastic differentiation – refractory anemia
Autoři:
Petra Vašeková 1; Peter Szépe 1,2; Ján Marcinek 1,2; Tomáš Balhárek 1,2; Lukáš Plank 1,2
Působiště autorů:
Konzultačné centrum bioptickej diagnostiky ochorení krvotvorby s celoslovenskou pôsobnosťou
Ústav patologickej anatómie Jesseniovej LF UK a UN Martin, Slovenská republika
1; Konzultačné centrum bioptickej diagnostiky ochorení krvotvorby s celoslovenskou pôsobnosťou
Martinské bioptické centrum, s. r. o., Martin, Slovenská republika
2
Vyšlo v časopise:
Vnitř Lék 2016; 62(9): 692-697
Kategorie:
Původní práce
Souhrn
Úvod:
Megaloblastová anémia (MA) je podtypom makrocytovej anémie, ktorý vzniká v dôsledku poruchy syntézy DNA. Jej častými príčinami sú deficity folátu a/alebo vitamínu B12. Najmiernejšie formy MA vedú len k vzniku makrocytózy bez sprievodnej anémie, ťažké formy až k vzniku súčasnej trombocytopénie a/alebo leukopénie. Stanovenie diagnózy MA by zväčša nemalo predstavovať závažnejší klinický problém, nevyhnutnosťou je však vylúčiť iné príčiny makrocytózy, vrátane myelodysplastického syndrómu (MDS).
Materiál a metodika:
V bioptickom registri sme v rokoch 2004–2015 identifikovali biopsie kostnej drene (KD) 126 pacientov s údajom o cytopénii/ách v periférnej krvi a súčasne vysloveným podozrením na MA resp. na MDS typu refraktérnej anémie (RA). Biopsie KD pacientov sme analyzovali so zameraním na parametre diferenciálnej diagnostiky, ako sú: (a) celková bunečnatosť KD a proporcionalita zastúpenia prekurzorov, (b) topografické pomery, (c) prítomnosť maturačných defektov a dysplastických zmien, (d) stupeň a rozsah myelofibrózy, (e) zásoby železitého pigmentu a (f) prítomnosti „zápalovej“ reakcie v KD. Na podporu histologickej analýzy boli použité imunohistochemické vyšetrenia.
Výsledky:
Súbor tvorilo 126 pacientov (61 mužov a 65 žien), priemerný vek bol 63 rokov (14–88 rokov). Anemický syndróm bol prítomný takmer u všetkých pacientov (121/126). Pacientov sme rozdelili do 3 podskupín – MA (31 pacientov), MDS-RA (39) a neklasifikovateľné prípady označené „DIF DG“ (56). Zmeny bunečnatosti KD sa vyskytli v 81 % a narušenie topografických pomerov v 73 % biopsií všetkých pacientov. Megaloblastovú prestavbu erytropoézy sme detekovali v 79 % všetkých biopsií, diagnostické dysplázie sme pozorovali v 25 % prípadov. Prstencové sideroblasty boli prítomné v 29 % všetkých biopsií. Poruchy lobulizácie a denzity jadier megakaryocytov sa vyskytovali u 61 % všetkých pacientov. V 14 % všetkých biopsií bola stróma KD bez myelofibrózy, len v 5 % vzoriek sa vyskytol stredne ťažký stupeň difúznej fibrózy. Stromálna „zápalová“ reakcia bola prítomná v 44 % všetkých prípadov. Zásoby železitého pigmentu absentovali v 26 %, resp. boli znížené v 35 % všetkých biopsií. Zvýšené zásoby boli prítomné v 33 % všetkých prípadov.
Záver:
Z pohľadu histopatológa je obtiažne odlíšiť zmeny krvotvorby v bioptickom obraze KD pacientov s MA a MDS-RA, keďže bioptické vyšetrenie umožňuje stanoviť jednoznačný a správny záver v približne 55 % prípadov. Zásadným problémom je rozhodnutie, či zmeny v KD sú alebo nie sú dôsledkom klonálneho ochorenia.
Kľúčové slová:
megaloblastová anémia – megaloblastová prestavba – refraktérna anémia
Úvod
Megaloblastová anémia (MA) sa považuje za podtyp makrocytovej anémie, ktorý vzniká v dôsledku poruchy normálnej syntézy DNA. Následkom je komplex zmien krvotvorných a iných tkanív, ako sú makrocytóza erytrocytov, abnormality leukocytov a trombocytov a epitelové zmeny najmä rýchlo sa deliacich buniek slizníc vrátane dutiny ústnej a tráviaceho systému [1]. Najmiernejšie formy MA vedú len k vzniku makrocytózy bez sprievodnej anémie, ťažké formy pri vzniku súčasnej trombocytopénie a/alebo leukopénie imitujú (pan-)cytopénie iného pôvodu [2]. Častými príčinami MA sú deficity folátu a/alebo vitamínu B12, pričom špecifickou formou MA je perniciózna anémia z nedostatku vitamínu B12 v dôsledku autoimúnnej atrofickej gastritídy [2,3].
Stanovenie diagnózy MA vo väčšine prípadov nepredstavuje závažnejší klinický problém, lebo pre určenie diagnózy obyčajne postačujú štandardné hematologické a biochemické vyšetrenia [4]. Z pohľadu patológa-bioptika možno dodať, že diagnózu MA z deficitu vitamínu B12 a/alebo folátu podporuje aj charakteristický morfologický obraz vo vyšetreniach tak náterov PK ako aj kostnej drene (KD). Súčasne je však potrebné vylúčiť iné príčiny makrocytózy vrátane myelodysplastického syndrómu (MDS), a to najmä typu refraktérnej anémie (RA) s výraznou megaloblastovou diferenciáciou. Do diferenciálne diagnostického algoritmu väčšiny pacientov sa trepanobioptické vyšetrenie KD obligatórne nezaraďuje. Odporúča sa však v prípadoch nejednoznačných laboratórnych výsledkov, najmä u pacientov s pancytopéniou nejasného pôvodu, a to na vylúčenie aplázie, myelodysplázie alebo inej neoplázie [5,6] a tiež u pacientov refraktérnych na substitučnú liečbu. Osobitné postavenie v diferenciálnej diagnostike týchto procesov nadobúdajú cytogenetické vyšetrenia, ktorých význam spočíva v dôkaze klonality MDS.
Skúsenosti aj na hematopatológiu špecializovaných patológov poukazujú na problémy a obtiažnosť odlíšenia zmien krvotvorby v bioptickom obraze pacientov s MA od zmien pri MDS. Preto sme sa v našej práci zamerali na zhodnotenie možností uvedenej diferenciálnej diagnostiky na základe vyšetrenia biopsie KD v korelácii s dostupnými klinickými a laboratórnymi údajmi, poskytnutými na sprievodnom lístku k biopsii.
Materiál a metodika
V bioptickom registri nášho pracoviska sme identifikovali trepanobiopsie KD 126 pacientov, vyšetrené v období január roku 2004 až december roku 2015, u ktorých bol uvedený údaj o cytopénii v jednom alebo viacerých krvných radoch a klinicky vyslovené podozrenie na MA, resp. na MDS typu RA. Išlo o biopsie KD, ktoré boli na naše pracovisko zaslané buď priamo z ambulancií hematologických pracovísk v Slovenskej republike (SR), alebo o konzultačné biopsie KD poslané na konzultačné vyšetrenie z iných pracovísk nášho odboru. Priamo zaslané trepanobiopsie boli v aktuálnom čase vyšetrenia vždy spracovávané štandardným spôsobom – po 24-hodinovej fixácii v 10% roztoku formaldehydu nasledovala 24-hodinová dekalcifikácia v chelatone III (EDTA – kyselina etylendiamintetraoctová) a odvodnenie v autotechnikone. Po zaliatí vzorky do parafínového bločku, resp. po získaní konzultačného bločku boli zhotovené 3–5 μm hrubé rezy a ofarbené panelom základných histologických a histochemických metód (HE, Giemsove farbenie, PAS reakcia, impregnácia podľa Gömöriho, Pearlsova reakcia, dôkaz α-naftol-AS-D-chloroacetátesterázy) a imunohistochemických (IHC) metód [dôkaz antigénov CD34, myeloperoxidázy, glykoforínu A (CD235A) a i. podľa potreby].
Všetky biopsie KD súboru 126 pacientov sme analyzovali retrospektívne so zameraním na hodnotenie:
- a) celkovej bunečnatosti drene (vo vzťahu k veku pacienta) a proporcionalitu relatívneho zastúpenia prekurzorov jednotlivých krvných radov
- b) topografických pomerov krvotvorby, resp. na ich patologickú disorganizáciu
- c) prítomnosť defektov v maturácii a prítomnosť dysplastických zmien v jednotlivých krvných radoch – v erytropoéze na črty megaloblastovej diferenciácie, nález dysplázií a na prítomnosť prstencových sideroblastov, v megakaryocytovom na kvantitatívne a kvalitatívne zmeny ako sú tendencia k zhlukovateniu, zmeny veľkosti, lobulizácie a denzity jadier
- d) stupeň a rozsah myelofibrózy
- e) zásoby železitého pigmentu a napokon aj na zhodnotenie
- f) prítomnosti „zápalovej“ reakcie strómy KD (prítomnosť lymfocytov, lymfoidných agregátov, plazmatických buniek, makrofágov a pod.)
Analýza IHC vyšetrení bola použitá na podporu histologickej analýzy – napr. expresia antigénov CD34 a MPO (spoločne s dôkazom chlóracetátesterázy) prispela k analýze granulopoézy (zastúpenia prekurzorov, maturačných pomerov a pod.) a expresia CD235A k zhodnoteniu zmien prekurzorov červeného radu. V prípade dostupných opakovaných biopsií pacienta sme porovnávali všetky nálezy. Nami zistené parametre sme porovnávali s klinicko-laboratórnymi údajmi uvedenými na sprievodnom liste k bioptickému vyšetreniu odosielajúcim klinikom.
Výsledky
Náš súbor tvorili biopsie KD 126 pacientov, z toho 61 (48 %) mužov a 65 (52 %) žien vo vekovom rozpätí 14–88 rokov, priemerný vek bol 63 rokov. Anemický syndróm rôzneho stupňa bol prítomný takmer u všetkých pacientov z nášho súboru (121 pacientov, tj. 96 %, u 4 pacientov neboli dostupné sprievodné klinické údaje a u 1 pacienta existoval len údaj o „poklese hemogramu“). Leukopénia sa vyskytovala v 66 (53 %) a trombocytopénia v 63 prípadoch (50 %). Vo viac ako polovici prípadov (73 pacientov, 58 %) bola anémia sprevádzaná aj makrocytózou erytrocytov a v 3 prípadoch aj hypersegmentáciou jadier neutrofilných leukocytov (tab. 1). Len v jedinom z vyšetrení 126 pacientov boli, aj to až pri rebiopsii pacienta uvedené odosielajúcim pracoviskom negatívne výsledky analýz FISH vyšetrení (bez prestavby génov MLL, RARA, ETV6, a negativita dôkazu anomálií 5. a 7. chromozómu).

Podľa výsledkov revidovanej histopatologickej diagnózy sme pacientov súboru rozdelili do 3 podskupín: na podskupinu 31 pacientov s MA, 39 s MDS-RA a na podskupinu 56 pacientov, tu označenú v skratke ako „DIF DG“ (problémové z hľadiska diferenciálnej diagnostiky), u ktorých nebolo možné na základe bioptického vyšetrenia rozhodnúť, či ide o klonálne ochorenie alebo reaktívne zmeny krvotvorby. Histomorfologické nálezy sú prehľadne zhrnuté v tab. 2, tu v stručnosti uvádzame ich prehľad a detailne len niektoré z nich.

Zmeny bunečnatosti KD sa vyskytli v 81 % všetkých prípadov: hypercelulárna dreň v 68 % všetkých prípadov, z toho u 77 % pacientov s MA, 56 % s MDS typu RA a u 71 % pacientov skupiny „DIF DG“. U 13 % všetkých pacientov bola KD hypocelulárna, a to najčastejšie pri RA (26 % všetkých) a menej často (3 %, resp. 9 %) v skupine MA, resp. „DIF DG“. V 19 % prípadov bola KD normocelulárna, približne rovnako často vo všetkých podskupinách pacientov.
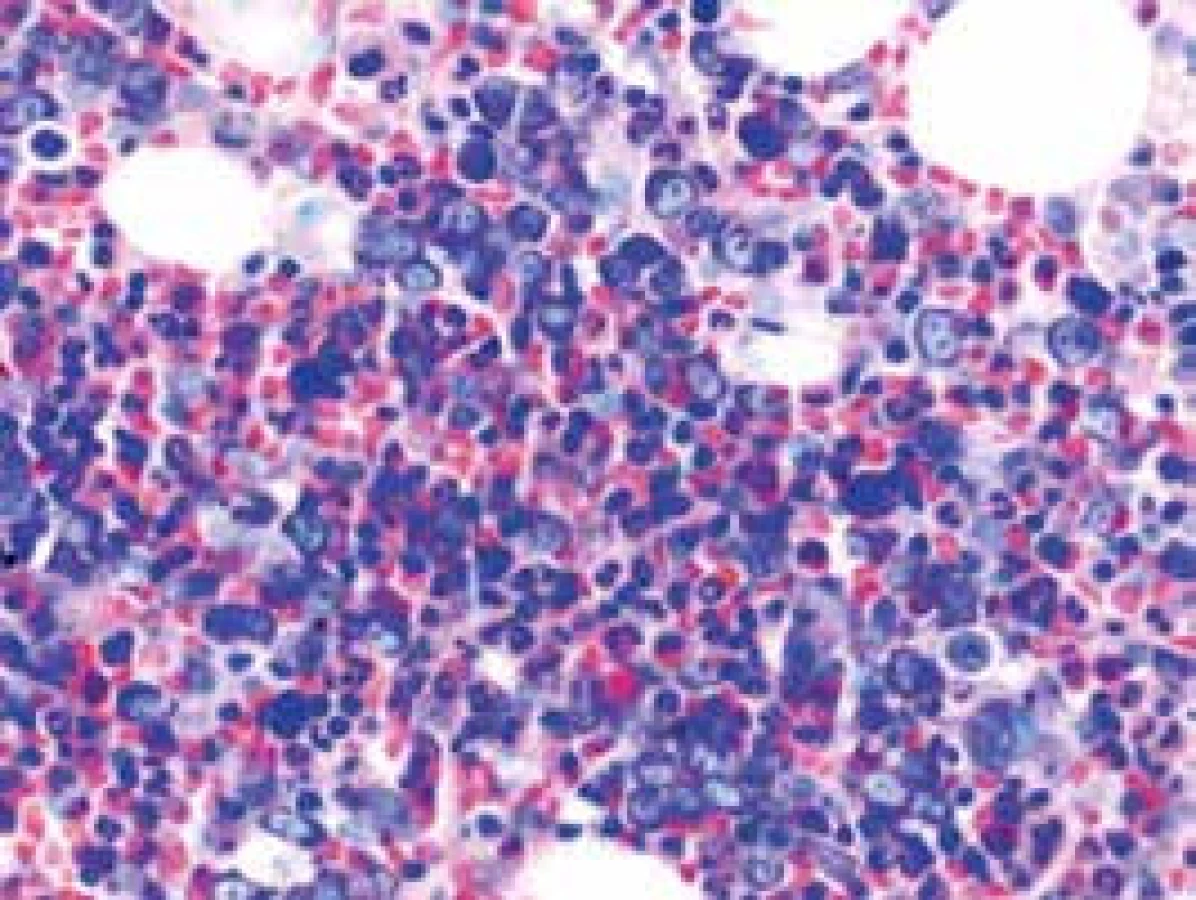
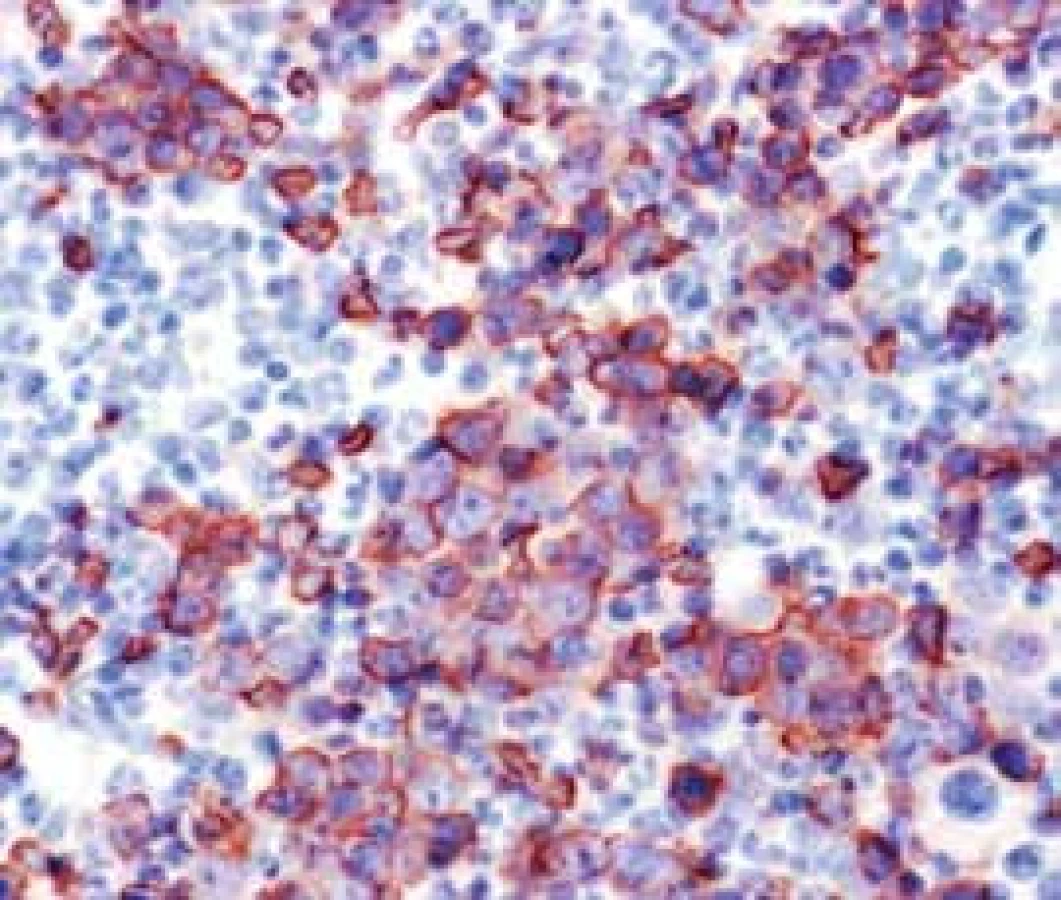
Narušenie pôvodných topografických pomerov sme identifikovali v 73 % všetkých pacientov, a to v 74 % prípadov MA, 92 % prípadov RA a u 59 % prípadov pacientov „DIF DG“. Megaloblastová prestavba erytropoézy (obr. 1 a 2) bola prítomná v 79 % všetkých biopsií KD, a to u 100 % prípadov MA, 95 % prípadov RA a v 57 % prípadov podskupiny „DIF DG“. Diagnosticky jednoznačné dysplastické zmeny (obr. 3) sme pozorovali v 25 % všetkých, a to v 16 % podskupiny MA, 46 % pacientov s RA a v 14 % prípadov „DIF DG“. Prstencové sideroblasty (obr. 4) boli prítomné v rôznom množstve v 29 % všetkých biopsií, najčastejšie (49 %) u pacientov s RA a menej často (13, resp. 25 %) u pacientov s MA, resp. podskupiny „DIF DG“. Na rozdiel od približne rovnakého výskytu kvantitatívnych zmien a tendencie k zhlukovateniu megakaryocytov vo všetkých sledovaných podskupinách sme zistili rozdiely v sledovaných kvalitatívnych parametroch. Poruchy lobulizácie a denzity jadier (obr. 2 a 5) sa vyskytli relatívne často, až u 61 % všetkých pacientov, a to najmä pacientov s RA (69 %) a podskupiny „DIF DG“ (68 %) a menej často (39 %) u pacientov s MA. V 14 % všetkých biopsií bola stróma drene bez myelofibrózy, v 41 % sme detekovali ľahký stupeň fokálnej a v 40 % difúznej fibrózy. Len v 5 % vzoriek sa vyskytol stredne ťažký stupeň difúznej fibrózy, a to výlučne u pacientov s MDS typu RA. Stromálna „zápalová“ reakcia bola prítomná v 44 % všetkých prípadov, bez podstatných rozdielov medzi prípadmi jednotlivých podskupín. Napokon zásoby železitého pigmentu úplne absentovali v 26 %, resp. boli znížené v 35 % všetkých biopsií. Zvýšené zásoby boli prítomné v 33 % všetkých prípadov, najčastejšie v skupine pacientov s RA (59 % biopsií pacientov s RA).



U 10 pacientov bolo v našom registri dostupné opakované bioptické vyšetrenie KD – u 6 pacientov s RA a u 4 z podskupiny „DIF DG“. Vo všetkých opakovaných biopsiách prípadov RA sa morfologický obraz nezmenil, v 4 prípadoch však išlo o prípady s pretrvávajúcou hypocelulárnou KD. U 2/4 pacientov podskupiny „DIF DG“ nálezy v rebiopsii favorizovali diagnózu MA, u 1 pacienta pretrvávali rozpaky nad možnosťou evolúcie MDS a u 4. bola opakovaná biopsia KD nereprezentatívna.
U jediného pacienta z nášho súboru, ktorý bol podľa klinických údajov na sprievodnom bioptickom lístku sledovaný a liečený pre pernicióznu anémiu bioptické vyšetrenie KD potvrdilo diagnózu MDS, a to typu RA.
Diskusia
Megaloblastová diferenciácia prekurzorov erytropoézy súvisí s poruchou syntézy DNA, ktorá môže mať rôzne príčiny. S výnimkou zriedkavých konštitučných geneticky podmienených ochorení sú najčastejšou príčinou megaloblastózy získané ochorenia, a to MA pri deficite vitamínu B12 a/alebo folátu, MDS, akútna myeloidná leukémia (najmä erytroidná) či užívanie niektorých liekov [7]. Správna diagnóza MA už počas včasných príznakov jej evolúcie, ako aj identifikácia jej etiopatogenézy sú pochopiteľne conditio sine qua non pre adekvátnu liečbu pacienta. Jej miernejšie formy sa môžu prejaviť len laboratórnou makrocytózou bez anémie, pričom makrocytové parametre erytrocytov sú charakterizované matematicky pomerne presne (MCV > 95 fl). V ťažších prípadoch MA už môže byť postihnutý aj biely a/alebo megakaryocytový rad [2]. Trepanobioptické vyšetrenie KD u pacientov s MA obyčajne nie je nutné, lebo správna diagnóza môže byť stanovená na základe klinických a laboratórnych hematologických vyšetrení (krvný obraz, náter periférnej krvi, stanovenie počtu retikulocytov, sérového bilirubínu, LDH, vyšetrenie metabolizmu železa, vitamínu B12 a folátu [5], prípadne aj gastroendoskopického vyšetrenia a vyšetrenia protilátok proti parietálnym bunkám žalúdočnej sliznice [3,4]. V časti prípadov s nejednoznačnou klinickou manifestáciou a nedostatočne diagnostickými laboratórnymi výsledkami je indikovaná biopsia KD [8] a patológ je konfrontovaný s náročnou úlohou diferenciálne diagnostickej položenej otázky: Ide o MA alebo o MDS typu RA? Odpoveď na túto otázku nie je vôbec jednoduchá.
Aj naša analýza ukázala, že MA obyčajne spôsobuje zmeny morfologického obrazu krvotvornej drene, a to nielen aktiváciu červeného radu so súčasnou megaloblastovou diferenciáciou, ale súčasne aj zvýšenie jej celkovej bunečnatosti. Zmeny v zmysle hypercelularity per se však nie sú špecifické pre MA, lebo hypercelularita bola prítomná približne rovnako často aj u pacientov podskupiny „DIF DG“. Podobné konštatovanie platí pre nález megaloblastovej erytropoézy, ktorá bola, okrem všetkých prípadov MA, vyznačená aj u 95 % (34/39 pacientov) s RA. Rovnako sme preukázali, že v súlade s literárnymi údajmi sa aj v časti prípadov MA, aj keď s nižšou incidenciou než pri RA, môžu vyskytnúť aj známky dysmegakaryopoézy. U časti pacientov s MA sa môže vyskytovať aj proliferácia granulopoézy s poruchou vyzrievania v zmysle hypersegmentácie jadier zrelých granulocytov, tieto poruchy sú však identifikovateľné takmer výlučne v cytologickom a nie bioptickom vyšetrení [7]. Známky topografickej disorganizácie, niekedy mylne považované za špecifické pre MDS, sa vyskytli až u troch štvrtín všetkých pacientov nášho súboru bez ohľadu na diagnózu, aj keď v zásade vždy v biopsii pacienta s RA a „len“ v troch štvrtinách pacientov s MA. Výhodné sa zdá byť posudzovať narušenie topografie prekurzorov krvotvorby v kontexte diagnosticky zrejmých dysplastických zmien a výskytu prstencovitých sideroblastov – všetky tieto zmeny sa totiž vyskytovali v značnej prevahe práve pri RA a menej často u pacientov s MA. Súčasne však platí aj opačné konštatovanie, že dysplastické zmeny nevylučujú diagnózu metabolicky podmienenej neklonálnej MA. Podobne aj nález pokročilejšej difúznej myelofibrózy v asociácii s výraznejšou stromálnou, pravdepodobne zápalovou reakciou, je nález favorizujúci diagnózu MDS typu RA pred záverom o MA. Keďže pri MDS všeobecne obyčajne dochádza k fibrotizácii interstícia KD rôzneho stupňa, k zvyšovaniu zásob železitého pigmentu, či k výraznejšej účasti zápalových buniek, tak uvedené znaky môžu predstavovať istú empirickú pomôcku pre odlíšenie MA od MDS typu RA. V zásade totiž pri MA obyčajne fibróza nie je prítomná, alebo je skôr fokálna a ľahká a zásoby železitého pigmentu sú zachované, resp. redukované, hoci uvedené neplatí univerzálne.
Z hľadiska diagnostiky (a pochopiteľne aj prognózy) pacientov so suspektným MDS je nevyhnutné poznať výsledky analýzy karyotypu pacienta, hoci v súčasnosti jediným cytogenetickým markerom ktorý definuje špecifický podtyp MDS predstavuje delécia 5q (11). Nám bol v aktuálnom čase bioptického vyšetrenia výsledok genetického vyšetrenia dostupný iba u jediného pacienta s MDS-RA. Aj keď nemožno vylúčiť oneskorenie výsledku genetických analýz (v čase odosielania biopsie nemusia byť ešte hematológovi dostupné), ide o zásadný problém, ktorého odstránenie môže prispieť k zvýšenou efektivity indikovaných bioptických vyšetrení.
Záver
Záverom možno zhrnúť, že diagnóza MA v biopsii KD je založená, podobne ako diagnostika iných ochorení krvotvorby, na princípe multiparametrického zhodnotenia viacerých tu diskutovaných histologických parametrov. Súčasne nemožno zabúdať, že rovnaké alebo prinajmenej podobné morfologické zmeny hemopoézy bývajú vyjadrené v bioptickom obraze v rôznom stupni tak pri MA, ako aj pri MDS, čo sťažuje bioptickú diferenciálnu diagnostiku. Podľa výsledkov našej analýzy v takmer 45 % prípadov makrocytovej anémie nemožno na základe histologického vyšetrenia vysloviť jednoznačný záver. Bioptická diagnostika MA je zložitá nielen pre prekrývanie morfológie, ale aj preto, že ide o algoritmus založený na subjektívnom hodnotení a závislý od skúseností diagnostikujúceho bioptika [9]. Preto aj klasifikácia WHO z roku 2008 [10] konštatuje, že zásadný problém pri hodnotení bioptických vzoriek predstavuje rozhodnutie, či prítomnosť opísaných zmien je alebo nie je dôsledkom klonálneho ochorenia. Stanovenie správnej diagnózy má zásadný terapeutický dopad, preto si vyžaduje multidisciplinárny a individuálny prístup k pacientovi, ktorý zohľadňuje a koreluje morfologické nálezy s klinickými, laboratórnymi či genetickými. Klonálne cytogenetické abnormality sú prítomné asi v polovici všetkých prípadov MDS a niektoré z nich sa považujú za „MDS-definujúce“. Z toho súčasne vyplýva, že negatívny cytogenetický výsledok nevylučuje diagnózu MDS [10]. Takisto platí, že genetické analýzy musia komplexné a potvrdené konvenčnou karyotypizáciou, nielen FISH alebo sekvenačnými technológiami [11].
Podporené projektom VEGA SR č. 1/0268 a projektami Centrum excelencie CEPV II (ITMS 26220120036), MBRKM a Biomed (ITMS 26220220187), ktoré sú riešené na JLF UK v Martine a spolufinancované z prostriedkov EÚ.
prof. MUDr. Lukáš Plank, CSc.
plank@jfmed.uniba.sk
Ústav patologickej anatómie JLF UK a UN Martin,
Slovenská republika
www.unm.sk
Doručeno do redakce 13. 4. 2016
Přijato po recenzi 28. 6. 2016
Zdroje
1. Gowda M, Shetty SA, Shetty VS. Megaloblastic anaemia. e-Journal of Dentistry 2014; 4(2): 608–620. Dostupné z WWW: <http://www.ejournalofdentistry.com/articles/e-JOD7BD5F6F930–02FB-1209D82056.pdf>.
2. Lequit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology 2010; 57(5): 655–670. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2559.2010.03612.x>.
3. Bizzaro N, Antico A. Diagnosis and classification of pernicious anemia. Autoimmunity reviews 2014; 13(4–5): 565–568. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2014.01.042>.
4. Roddie C, Davis B. Iron, B12 and folate. J Medicine 2009; 37(3):125–128. Dostupné z DOI: <http://dx.doi.org/10.1016/j.mpmed.2008.12.009>
5. Parker-Williams E. Investigation and management of anaemia. Medicine 2004; 32(5): 14–20. Dostupné z DOI: <http://dx.doi.org/10.1383/medc.32.5.14.33954>.
6. Čermák J. Myelodysplastický sydrom. Pokroky v diagnostice a léčbe během 30 let trvání registru nemocných s myelodysplastickým syndromem v ÚHKT. Vnitř Lék 2012; 58(Suppl 2): 2S8–2S15.
7. Foucar K, Viswanatha DS, Wilson CS. Non-neoplastistic disorders of bone marrow. First series. Fascicle 6. ARP Press: Washington DC 2008 : 84–89. ISBN 978–1-933477–04–6.
8. Čermák J. Trombocytopenie u myelodysplastického syndromu. Vnitř Lék 2010; 56(Suppl 1): S39-S42.
9. Garcia-Manero G. CME Information: MDS: 2014 Update on diagnosis, risk-stratification and management. Am J Hematol 2014; 89(1): 97–108. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.23642>.
10. Swerdlow SH, Campo E, Harris NL et al. World Health Organization Classification of Tumours of Hematopoetic and Lymphoid Tissues. 4th ed. IARC Press: Lyon 2008 : 86–103. ISBN 9789283224310.
11. Arber DA, Orazi A, Hasserijan R et al. The 2016 revision to the World Health Organisation classification of myeloid neoplasms and acute leukemia. Blood 2016; 127(20): 2391–2405. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2016–03–643544>.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2016 Číslo 9
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Arytmogenní kardiomyopatie levé komory
- Jak postupovat při podezření na sekundární arteriální hypertenzi
-
Schnitzlerové syndrom
Diferenciální diagnostika, přehled léčebných možností a popis 5 případů léčených anakinrou - Perorálne antikoagulanciá v primárnej a sekundárnej prevencii vénovej tromboembólie