
IgA pemfigus provázející mnohočetný myelom vymizel při léčbě bortezomibem (Velcade), cyklofosfamidem a dexametazonem. Popis případu a přehled literatury
IgA pemphigus accompanying multiple myeloma has disappeared following the treatment with bortezomib (Velcade), cyclophosphamide and dexamethasone. Case study and literature review
IgA pemphigus, resembling subcorneal pustulous dermatosis, represents a rare complication of IgA type monoclonal gammopathy. The patient dates the onset of initial symptoms of vesicular ‑ bullous disease to 1990. She was first examined at our clinic in 2001 with the following conclusion “type IgA monoclonal gammopathy of unknown significance”. The first immunosuppressive treatment of vesicular - bullous disorder was administered in 2003 (dexamethasone 20 mg on days 1 – 4 and 15 – 18 in monthly cycles + daily cyclophosphamide 50 mg). Cyclophosphamide was administered for 6 months in total and dexamethasone for further 3 months. During the treatment, intensity of the skin disorder ameliorated and monoclonal IgA levels decreased to non‑detectable levels. Nevertheless, skin symptoms recurred immediately after dexamethasone treatment in its original intensity was terminated, even though the concentration of monoclonal immunoglobulin IgA remained below the sensitivity of quantitative detection for further 6 months (positive immunofixation only). Six rituximab 600 mg infusions were administered in a weekly interval after stopping cyclophosphamide and dexamethasone to prevent early recurrence of skin symptoms but this treatment was without any lasting effect. Transformation into multiple myeloma was identified in 2007. First line treatment (cyclophosphamide, adriamycin and dexamethasone – CAD) remained without any haematological or dermatological treatment response. Second line treatment (thalidomide, cyclophosphamide and dexamethasone – CTD) brought about significant deterioration of skin symptoms up to the clinical picture of erythrodermia. Third line treatment (bortezomib 1.3 mg/ sqm i.v. on days 1, 4, 8 and 15, cyclophosphamide 50 mg daily and dexamethasone 20 mg on days 1 – 4 and 15 – 18 in 28-day cycles – VCD) resulted in rapid decline in monoclonal immunoglobulin IgA concentrations immediately following the first cycle and to negative immunofixation after 5 cycles. In total, six VCD cycles were administered. The patient has had no skin symptoms from the third cycle of this treatment and complete skin and haematological remission has been maintained for 12 months after completion of bortezomib - containing treatment. Combined treatment containing bortezomib has proven useful in the treatment of IgA pemphigus accompanying monoclonal gammopathy of uncertain significance transformed into multiple myeloma.
Key words:
IgA pemphigus – subcorneal pustulous dermatosis – bortezomib – rituximab – monoclonal gammopathy of uncertain significance – multiple myeloma
Autoři:
Z. Adam 1; J. Feit 2; M. Krejčí 1; L. Pour 1; V. Vašků 3; Z. Čermáková 4; J. Mayer 1; R. Hájek 1
Působiště autorů:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Vorlíček, CSc.
1; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MU Dr. Josef Feit, CSc.
2; I. dermatovenerologická klinika Lékařské fakulty MU a FN u sv. Anny Brno, přednosta doc. MU Dr. Vladimír Vašků, CSc.
3; Oddělení klinické biochemie Fakultní nemocnice Brno, pracoviště Bohunice, přednosta doc. MU Dr. Milan Dastych, CSc.
4
Vyšlo v časopise:
Vnitř Lék 2009; 55(10): 981-990
Kategorie:
Kazuistiky
Souhrn
IgA pemfigus podobný subkorneální pustulózní dermatóze je vzácně se vyskytující komplikací monoklonální gamapatie typu IgA. Pacientka datuje vznik prvních příznaků vezikulobulózního onemocnění do roku 1990. V roce 2001 byla poprvé vyšetřena na našem pracovišti se závěrem „monoklonální gamapatie nejasného významu typu IgA“. První imunosupresivní léčba pro vezikulobulózní onemocnění byla podána v roce 2003 (dexametazon 20 mg 1. – 4. den a 15. – 18. den v měsíčních cyklech + denně cyklofosfamid 50 mg). Cyklofosfamid byl podáván celkem 6 měsíců, dexametazon o 3 měsíce déle. Po dobu léčby se snížila intenzita kožního onemocnění a poklesl monoklonální IgA pod detekovatelné hodnoty. Kožní projevy však ihned recidivovaly po přerušení dexametazonu v původní intenzitě, i když koncentrace monoklonálního imunoglobulinu IgA zůstala po dalších 6 měsíců pod citlivostí kvantitativní detekce (pouze pozitivní imunofixace). Pro časnou recidivu kožních projevů po ukončení cyklofosfamidu a dexametazonu bylo podáno 6 infuzí rituximabu 600 mg v týdenních intervalech, ale bez přetrvávajícího efektu. V roce 2007 zjištěna transformace v mnohočetný myelom. Léčba první linie (cyklofosfamid, adriamycin a dexametazon – CAD) byla bez hematologické a kožní léčebné odpovědi. Léčba druhé linie (thalidomid cyklofosfamid a dexametazon – CTD) výrazně zhoršila kožní projevy až do obrazu erytrodermie. Léčba třetí linie (bortezomib 1,3 mg/ m2 i.v. 1., 4., 8. a 15. den, cyklofosfamid 50 mg denně a dexametazon 20 mg 1. – 4. den a 15. – 18. den ve 28denních intervalech – VCD) vedla k prudkému poklesu koncentrace monoklonálního imunoglobulinu IgA ihned po prvním cyklu a k negativní imunofixaci po 5 cyklech. Celkem bylo podáno 6 cyklů VCD. Od třetího cyklu této léčby je paní zcela bez kožních projevů a kompletní kožní i hematologická kompletní remise trvala při poslední kontrole již 12 měsíců po ukončení bortezomib obsahující léčby. Kombinovaná léčba obsahující bortezomib se osvědčila při léčbě IgA pemfigu provázejícího monoklonální gamapatii nejistého významu, která se transformovala v mnohočetný myelom.
Klíčová slova:
IgA pemfigus – subkorneální pustulózní dermatóza – bortezomib – rituximab – monoklonální gamapatie nejistého významu – mnohočetný myelom
Úvod
Klonální proliferace plazmocytů tvoří ve většině případů kompletní molekulu monoklonálního imunoglobulinu, asi u 1/3 nemocných jsou tvořeny pouze volné lehké řetězce, případně jejich fragmenty. Jen výjimečně je dediferenciace tak značná, že není tvořen žádný monoklonální imunoglobulin či jeho části [1–6].
Poškození organizmu monoklonálním imunoglobulinem lze rozdělit na:
- fyzikální (projevy zvýšené viskozity, projevy kryglobulinemie)
- projevy způsobené reakcí monoklonálního imunoglobulinu s antigeny vlastního těla
- depozity volných lehkých řetězců v amyloidové či neamyloidové podobě
Monoklonální imunoglobuliny se na-štěstí ve většině případů nevážou na žádné tělu vlastní antigeny, až na vzácné výjimky, kdy navázání monoklonálního imuglobulinu na tělu vlastní antigen způsobí poškození imunitním mechanizmem.
Jedním z těchto výjimečných projevů monoklonální gamapatie je vesikulopustulózní kožní onemocnění, způsobené depozity monoklonálního imunoglobulinu typu IgA v epidermis. Dermatologická terminologie těchto vezikulopustulózních kožních onemocnění není jednotná. Nejčastěji používaným termínem pro tyto formy kožního postižení je IgA pemfigus podobný subkorneální pustulózní dermatóze.
V literatuře lze nalézt široké spektrum synonym, které se objevilo od popsání prvního případu [7,8]: intraepidermální IgA pustulosis [9], IgA pemfigus [10], intercelulární IgA dermatóza [11] a intercelulární IgA vezikulopustulózní dermatóza [12]. Ve všech případech byla imunofluorescenčně prokázána depozita imunoglobulinu IgA. V některých případech šlo o samostatné onemocnění, v jiných případech souvisely kožní projevy s IgA monoklonální gamapatií [13].
Ongenae (1999) navrhl zastřešující termín intercelulární IgA dermatóza připomínající IgA pemfigus pro všechny vezikulopustulózní projevy s IgA depozity v dermis [14]. Nicméně nejčastějším termínem, používaným v publikovaných popisech, je právě IgA pemfigus podobný subkorneální pustulózní dermatóze. V popisech těchto případů je uváděno pouze částečné zlepšení při dostupné léčbě. Domníváme se, že toto je první popis případu, kdy při léčbě bortezomibem v kombinaci s dalšími léky vymizely dlouhodobě kožní projevy i monoklonální imunoglobulin.
Popis případu
Vstupní vyšetření monoklonální gamapatie v roce 2001
Žena narozená roku 1940 byla poprvé vyšetřena na našem pracovišti v srpnu roku 2001 ve věku 61 let. Při vstupním vyšetření udala, že již nejméně 10 let, tedy od roku 1990, má kožní onemocnění, které bylo nazýváno pracovně pemfigem. Na trupu a končetinách se jí tvořila vezikulózní ložiska, která postupně zasychala, puchýřky praskaly a následně se tvořila pigmentovaná ložiska. Na sliznicích tyto projevy neměla. V dokumentaci z kožního vyšetření z roku1996 je závěr patologa: Jedná o epidermis s mírně zvýšeným, od spodiny se odlučujícím stratum corneum, mezi stratum corneum a stratum spinosum jsou úsekovitě zbytky puchýřků. Stratum granulosum je 5–7 vrstev vysoké, rovněž bazofilně se přibarvující, akantóza, místy spongióza. V horní dermis jsou husté, ale nepříliš časté lymfocytární perivaskulární infiltráty. Toto první vyšetření z roku 1996 bylo ukončeno konstatováním, že se může jednat o impetigo, dále o subkorneální pustulózní dermatózu, event. IgA pemfigus.
Při prvním vyšetření na našem pracovišti v srpnu roku 2001 pacientka udávala zažívací potíže, jejichž příčina byla již dříve uzavřena jako dráždivý tračník. Byla po hysterektomii pro myomatózní uterus. Kožní lékaři pacientku v rámci vyšetřování bulózního onemocnění odeslali na imunologii, kde prokázali v séru monoklonální imunoglobulin typu IgA λ, a to byl důvod odeslání na hematoonkogické pracoviště. Bolesti kostí neměla, dušnost a teploty také ne. Mimo pemfigu ji trápila jedině zvýšená únavnost.
V objektivním klinickém nálezu byl patologický pouze kožní nález, drobné vezikuly, které postupně praskaly, a obnažená spodina se hojila pigmentovanými jizvami. V ústech tyto projevy pacientka neměla.
Vstupní hematologické vyšetření prokázalo monoklonální imunoglobulin IgA λ v séru v koncentraci 18,80g/l. Proteinurie dosahovala pouze 0,09g/l, ale v moči bylo přítomno stopové, nekvantifikovatelné množství volných λ řetězců. V myelogramu ze sternální punkce bylo 7,2–10% plazmatických buněk. β2 mikroglobulin byl nízký – 1,18mg/l. Hodnoty krevního obrazu byly v normě, leukocyty 5,2 × 109/l, erytrocyty 4,18 × 1012/l, trombocyty 170 × 109/l. Hodnoty urey a kreatininu byly v normě a RTG snímky skeletu neprokázaly patologické změny typické pro mnohočetný myelom. Kvantitativní koncentrace imunoglobulinů byla: IgG 7,07g/l (norma 7–16g/l), IgM 0,55g/l (norma 0,4–2,30g/l), IgA 6,96g/l (norma 0,7–4,0g/l). CT mediastinálních a břišních uzlin neprokázalo lymfadenopatii. Vyšetření hrudní a bederní páteře magnetickou rezonancí neprokázalo změny svědčící pro mnohočetný myelom. Měření kostní denzity metodou DEXA prokázalo jenom osteopenii, snížení na hodnoty –1,5 SD.
Toto standardní diferenciálně diagnostické vyšetření monoklonální gamapatie jsme tedy uzavřeli jako monoklonální gamapatie nejistého významu při pemfigu s doporučením pravidelných kontrol.
Opakovaná dermatologická vyšetření
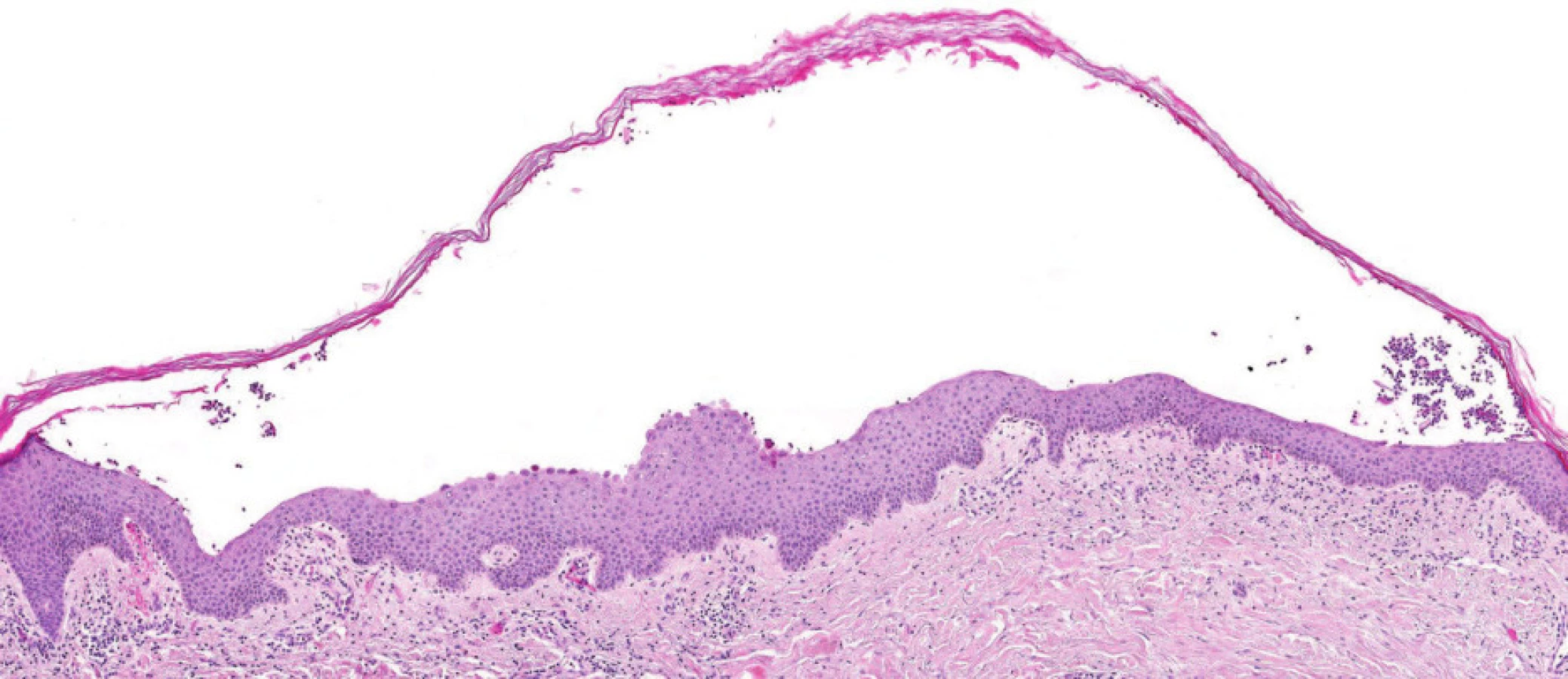
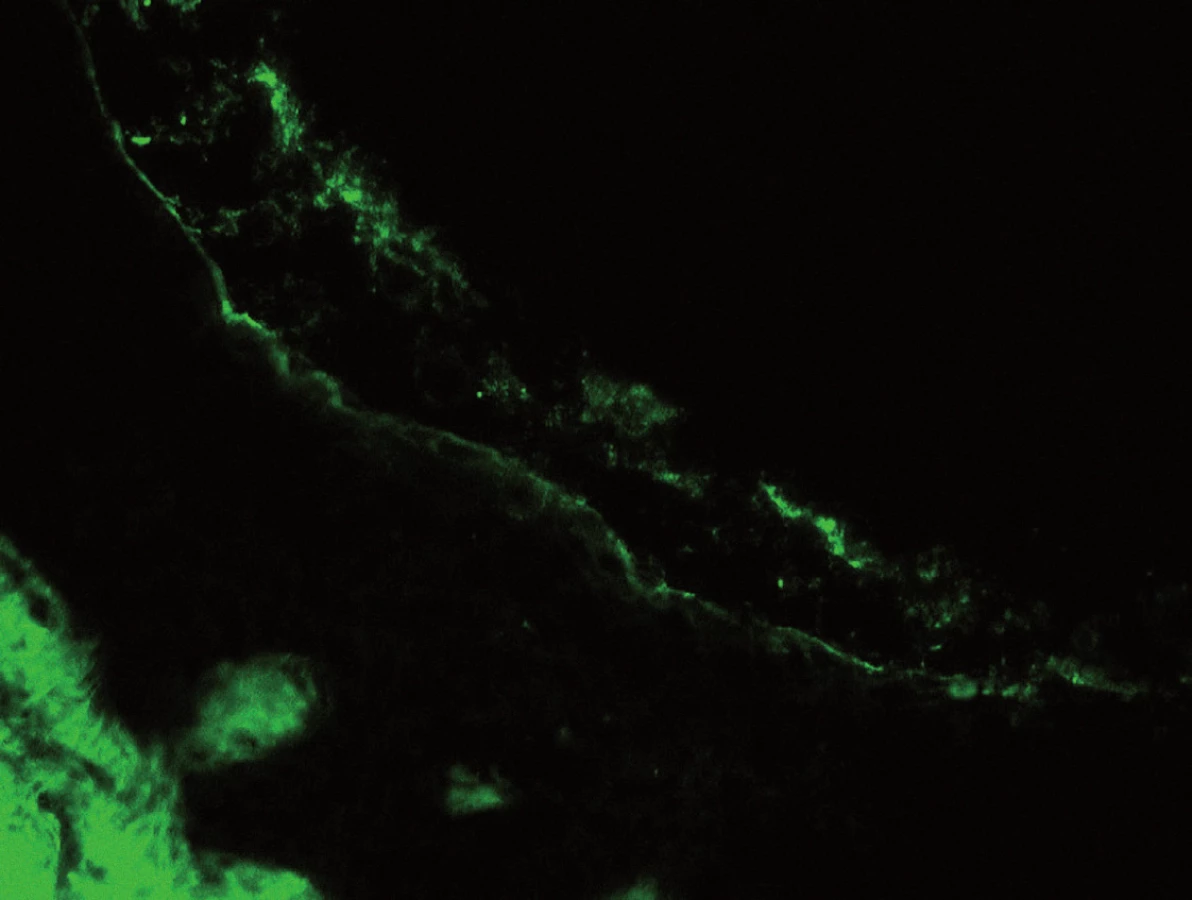
Poslední histologické vyšetření kůže bylo provedeno v roce 2007, kdy patolog jednoznačně uzavírá: Epidermis se subkorneálním puchýřkem. Puchýřek obsahuje jen malé množství buněk, jedná se o neutrofily. Ve spodině puchýřku jsou patrny zakulacené akantolytické buňky, jen mírná spongióza. Eozinofily jsou přítomny jen minimálně. V preparátu nebyly vůbec zastiženy plazmatické buňky. Přímá imunofluorescence: ve spodině buly je pozitivita IgA s mírným pronikáním mezi buňky epidermis. Dále je mírná pozitivita barvení na fibrin perivaskulárně a subepidermálně. Nález odpovídá IgA pemfigu připomínajícímu subkorneální pustulózní dermatózu (obr. 1–4).


Kontrolní hematologické vyšetření v roce 2002
V rámci kontrolního hematologickéhovyšetření v srpnu roku 2002 bylo zopakováno vyšetření kostní dřeně trepanobiopsií s histologickým hodnocením válečku. Bylo nalezeno 7,5% plazmatických buněk dominantně exprimujících λ řetězce. Cytologické hodnocení roztěru aspirátu kostní dřeně uvádí 9,6% plazmocytů. Koncentrace monoklonálního imunoglobulinu IgA se snížila na 11,50g/l. Cytogenetické vyšetření v roce 2004 prokázalo monozomii 13. chromozomu. Závěr vyšetření byl stejný jako předchozí – monoklonální gamapatie nejistého významu typu IgA λ.
V únoru roku 2003 již byl jištěn pokles polyklonálních imunoglobulinů, IgG 4,80g/l, IgM 0,37g/l, IgA 6,06g/l. Monoklonální IgA zůstával v nezměněné koncentraci 12g/l. Snížení polyklonálních imunoglobulinů splňoval jedno malé kritérium, přítomnost monoklonálního imunoglobulinu druhé malé kritérium dle Durieho a Salmonových kritérií. Počet plazmocytů v kostní dřeni byl na hranici 10%, nebyly však žádné kostní změny, anémie či renální insuficience. PET zobrazení neprokázalo patologická ložiska zvýšení konzumpce glukózy. Stav jsme hodnotili jako hraniční stav mezi monoklonální gamapatií nejistého významu a asymptomatickým myelomem.
Imunosupresivní léčba pemfigu
Léčba IgA pemfigu cyklofosfamidem a dexametazonem v roce 2003
Vzhledem k tomu, že vezikulobulózní onemocnění bylo aktivní a pacientka tím velmi trpěla, byla v březnu roku 2003 zahájena po domluvě s dermatologem léčba cyklofosfamidem 2krát 50mg denně a prednisonem. Po měsíci této léčby však nedošlo ke zlepšení kožních změn, proto byl prednison zaměněn za dexametazon 20mg p.o. 1.–4. den a 15.–18. den v měsíci. Cyklofosfamid pacientka užívala po dobu 6 měsíců – od března do srpna roku 2003. Pak bylo podávání cyklofosfamidu ukončeno. Důvodem k ukončení byl výsledek stanovení monoklonálního Ig, v době ukončení byl zastaven pokles monoklonálního IgA imunoglobulinu.
Dále byl ponechán pouze dexametazon 20mg po 4 dny ve 14denních intervalech. V době mezi dávkami dexametazonu paní užívala 10–20mg prednisonu. Důvodem k ponechání kortikoidů byl jejich příznivý efekt na výsevy morf IgA pemfigu.
Od zavedení dvou vysokých dávek dexametazonu měsíčně se intenzita aktivity pemfigu výrazně zmenšila, ale nevymizela úplně. V době přerušení cyklofosfamidu ještě byl detekovaný monoklonální imunoglobulin, ale překvapující bylo, že od ledna roku 2004 po sedm následujících měsíců poklesl monoklonální imunoglobulin na stopové množství pod možností kvantitativní detekce, ale s pozitivním průkazem imunofixační elektroforézou.
Aktivita IgA pemfigu byla při výše uvedené léčbě snížena, přesto se občasné výsevy puchýřků objevovaly. V lednu roku 2004 jsme přerušili i podávání uvedených vysokých dávek dexametazonu. Po relativním klidu v měsících únoru a březnu roku 2004 se v dubnu roku 2004 aktivita vezikulobulózního onemocnění opět zvýšila, a to v době, kdy koncentrace monoklonálního IgA stále zůstávala pod hranicí kvantitativní detekce.
Léčba IgA pemfigu antiCD20 protilátkou, rituximabem v roce 2004
Vzhledem k četným popisům pozitivního vlivu protilátky antiCD20 na pemfigus jsme chtěli pacientce touto léčbou ulevit od stálého utrpení, které ji puchýřnaté onemocnění připravilo.
Od května roku 2004, tedy v době, kdy měla nekvantifikovatelnou koncentraci monoklonálního imunoglobulinu, jsme začali s léčbou protilátkou antiCD20 – rituximab (Mabthera). Pacientka od 25. 5. 2004 do 29. 6. 2004 dostala celkem 6 infuzí v dávce 600mg rituximabu. Po dobu infuzí byla pacientka bez potíží, aktivita pemfigu se výrazně zmenšila, ale za 4 týdny od poslední infuze rituximabu došlo opět k podstatnému zhoršení pemfigu, takže jsme se museli vrátit k standardní léčbě dexametazonem 20mg 2 dny po sobě, vždy 1. a 2. a pak 15. a 16. den v měsíci. Ve dnech, kdy neměla dexametazon, užívala 10–20mg prednisonu.
Pacientka byla dále ve sledování u kožních specialistů, ale v postatě nic jiného než kortikoidy její potíže nezmírnilo.
Přechod do symptomatického myelomu v roce 2007
Monoklonální imunoglobulin se začal od července roku 2004 postupně zvyšovat a v březnu roku 2007 poprvé přestoupil hodnotu 20g (20,4g/l) a v moči se objevilo vyšší množství bílkoviny (0,77g/l a z toho 0,48g/l lehkých řetězců) (tab. 1). Zároveň se zhoršovaly i kožní projevy při zavedené léčbě.

Proto v říjnu roku 2007 bylo provedeno další vyšetření kostní dřeně, v trepanobiopsii bylo nalezeno 31–50% plazmocytů, cytologicky jich bylo přítomno jen 28,8%. Monoklonální imunoglobulin dosáhl hladiny 28g/l a proteinurie se pohybovala v posledních měsících před trepanobiopsií mezi 0,5 a 2,57g/l a denzitometrie monoklonálního imunoglobulinu 0,32–1,56g/l v moči.
Jednalo se tedy o jasné známky progrese v mnohočetný myelom dle zvýšení koncentrace monoklonálního imunoglobulinu v séru i v moči. Na RTG snímku skeletu byla zřetelná nová patologická fraktura těla Th7 a L2. FDG-PET zobrazení prokázalo pouze drobné ložisko vyšší konzumpce glukózy v levém rameni.
Léčba mnohočetného myelomu kombinací CAD (cyklofosfamid, adriamycin, dexametazon)
Tyto jednoznačné důkazy přechodu asymptomatického mnohočetného myelomu v symptomatický byly indikací k zahájení léčby chemoterapií CAD (cyklofosfamid 500mg/m2 i.v. 1. a 15. den i.v., doxorubicin 9mg/m2 i.v. 1.–4. dena dexametazon 40mg p.o. 1.–4. a 15. až18. den cyklu). Léčba byla zahájena v říjnu roku 2007.
Po 4 cyklech byla v březnu roku 2008vyhodnocena léčebná odpověď. Monoklonální imunoglobulin IgA poklesl pouze nepatrně z 27,7g/l na 20g/l, odpad lehkých řetězců močí se prakticky nezměnil, klesl z 0,72g/l na 0,60g/l. Bylo tedy dosaženo jen minimální léčebné odpovědi. Co se týče vezikulobulózního onemocnění, vždy v době podání dexametazonu ustaly nové výsevy puchýřků a po jeho ukončení se vždy začaly postupně obnovovat, došlo tedy k mírnému a dočasnému zlepšení IgA pemfigu.
Léčba mnohočetného myelomu kombinací CTD (cyklofosfamid, thalidomid, dexametazon)
Pro neuspokojivou léčebnou odpověď byla v březnu roku 2008 zahájena chemoterapie CTD (cyklofosfamid tbl. 50mg denně, thalidomid – Myrin – 100mg večer a dexametazon 20mg kapsle 1.–4. den a 15.–18. den). Profylakticky byl proti hluboké žilní trombóze k této léčbě podáván Fragmin 5 000 j s.c. a proti pneumocystové pneumonii Sumetrolim 480mg denně. Po měsíci této léčby se však výrazně zhoršilo kožní vezikulobulózní onemocnění, které v půli dubna roku 2008 dosáhlo formy erytrodermie s nutností hospitalizace. Proto bylo uvedené léčebné schéma CTD po 6 týdnech (1,5 cyklu) ukončeno a dále léčeny lokálně kožní projevy. Po ukončení podávání thalidomidu erytrodermie ustoupila. Moklonální IgA zůstal na stabilní hodnotě 19,3g/l, takže žádná léčebná odpověď.
Léčba mnohočetného myelomu kombinací VCD (bortezomib, cyklofosfamid, dexametazon)
Dne 6. 5. 2008 (po pauze nutné ke zvládnutí kožních projevů indukovaných thalidomidem) byla zahájena chemoterapie VCD, bortezomib (Velcade) 1,3mg/m2 i.v. 1., 4., 8. a 15. den, cyklofosfamid 500mg/m2 i.v. 1. a 15. den a dexametazon 40mg p.o. 1.–4. a 15. až18. den.
Koncentrace monoklonálního Ig bylav době zahájení chemoterapie 19,3g/l v séru a 0,48g/l v moči. Po prvním cyklu, při zahájení druhého cyklu (3. 6. 2008) se prudce snížila koncentrace monoklonálního IgA na kvantitativně nedekovatelnou hodnotu a proteinurie poklesla na hodnoty 0,10g/l se stopovým množstvím volných lehkých řetězců (pozitivní imunofixační elektroforéza). Monoklonální imunoglobulin IgA zůstal od června roku 2008 již pod možností kvantitativní detekce, ale jeho nízká koncentrace byla stále prokazatelná imunofixační elektroforézou v séru. Při zahájení 6. cyklu chemoterapie VCD však již byla i imunofixační elektroforéza negativní.
Vezikulobulózní kožní projevy se po1. cyklu výrazně zmenšily a od 3. cyklujiž nedocházelo k výsevu nových puchýřů. Na kůži jsou patrné pouze jizvy po této nemoci, bez jakéhokoliv nového výsevu. Chemoterapie byla ukončena po 6. cyklu a dále v ní nebylo pokračováno z důvodu postupně narůstajících neuropatických potíží projevujících se celkovou slabostí. Neuropatie je běžným nežádoucím účinkem léčby bortezomibem.
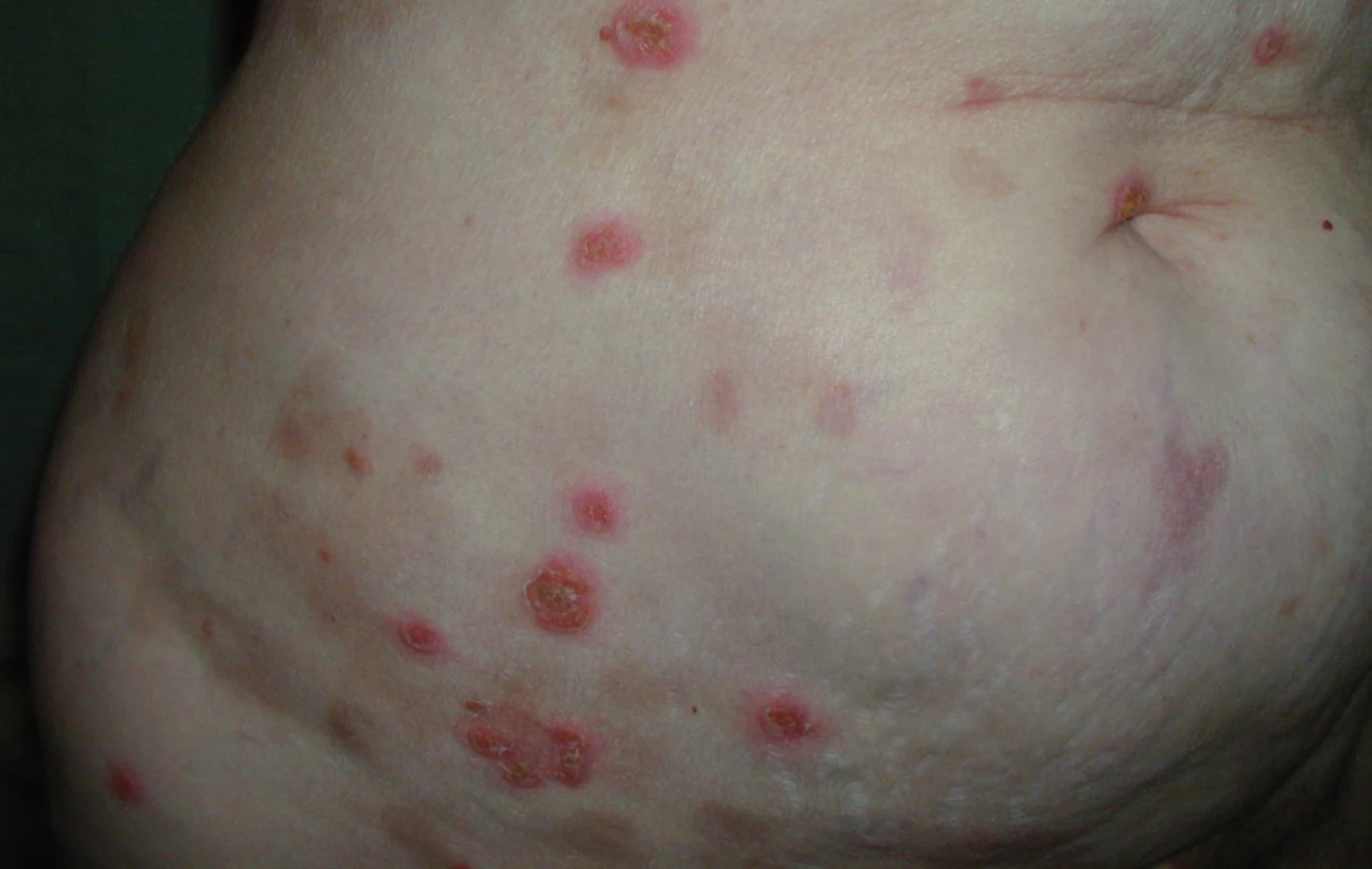
Při poslední kontrole v říjnu roku2009 (12 měsíců od ukončení léčby)zůstává pacientka zcela bez projevů vezikulobulózního onemocnění, probíhají pouze pravidelné aplikace ibandronátu a pacientka od ukončení léčby nemá žádné kortikoidy. Stav hodnotíme jako kompletní remisi nemoci s negativní imunofixací a normálním krevním obrazem. Vývoj některých laboratorních parametrů, léčbu a aktivitu vezikulobulózních kožních projevů zachycuje tab. 1. Kožní projevy nemoci před zahájením léčby obsahující bortezomib ukazují obr. 5 a 6, kůže bez těchto projevů pak obr. 7 a 8.



Diskuze
Monoklonální gamapatie může míti kožní projevy. Shrnujeme je v tab. 2 [15–17]. Jde o výjimečné komplikace monoklonální gamapatie, které zřejmě kvůli své vzácnosti nejsou včas rozpoznávány. Možná i proto, že internista či hematolog si bez obrázkové učebnice obtížně představuje, jaké kožní onemocnění se pod tím či oním dermatologickým termínem ukrývá.
![Spektrum kožních projevů monoklonálních gamapatií [15,17].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/99858f238c80a383e31a92fdcd7079ef.png)
Jedním z výjimečných onemocnění je chronicky relabující vezikulobulózní onemocnění provázející, či dokonce předcházející monoklonální gamapatii typu IgA, nazývané IgA pemfigem podobným subkorneální pustulózní dermatóze [16,18–25]. Může být kombinované s dalšími kožními projevy, např. pyoderma gangrenosum [26–28] nebo s kryoglobulinemií a nemocí chladových aglutininů [29].
Pacienti trpí vezikulobulózní erupcí, která postihuje trup a končetiny, ale i podpaží, třísla a krk. Subkorneální pustulózní dermatóza zpravidla nepostihuje ústní sliznici ani další mukózní povrchy na rozdíl od klasického paraneoplastického pemfigu, u něhož dochází k poškození nejen ústní sliznice, ale i bronchiální výstelky [30–34].
Zcela výjimečný je popis výsevu puchýřů v místě aplikace G CSF [35] anebo v souvislosti s léčebnou aplikaci interferonu α [36] u pacientů s monoklonální gamapatií.
Pro vezikulobulózní onemocnění s prokazatelnými depozity IgA imunoglobulinu při imunofluorescenčním vyšetření je navrženo skupinové označené intercelular IgA dermatosis [14], které lze použít pro celou skupinu kožních puchýřnatých onemocnění s IgA pemfigem (klinicky a histologicky jako pemfigus) na jedné straně a intercelulární vezikulopustulózní dermatózou či subkorneální pustulózní dermatózu s IgA depozity na straně druhé.
V literatuře je možné nalézt velké spektrum synonym: intraepidermální Ig pustulosis [9], IgA pemfigus [10], intercelulární IgA dermatóza [11] a intercelulární IgA vezikulopustulózní dermatóza [12]. Ve všech těchto případech byla imunofluorescenčně prokázána depozita imunoglobulinu IgA. Ve většině případů byla přítomna monoklonální gamapatie typu IgA, méně často šlo o samostatné onemocnění [25,37–39].
Patofyziologickým pokladem je vazbaimunoglobulinu (někdy zřejmě monoklonálního imunoglobulinu IgA) na různé antigeny kůže. Velmi častá je souvislost s monoklonální gamapatií, dále pak byla popsána souvislost s autoimunitním zánětlivým onemocněním střeva, s revmatoidní artritidou či erythema gangrenosum [40–51]. Drobné rozdíly v klinické formě nemoci zřejmě souvisejí se zaměřením antigenní determinanty deponovaného IgA gamaglobulinu. V některých případech byla antigenní determinanta monoklonálního IgA zaměřena na klasický pemfigový antigen, ale byly popsány i jiné kožní antigeny, proti nimž byl zaměřen imunogloubulin typu IgA. Monoklonální imunoglobulin nemusí být detekován zpočátku v době vzniku kožních potíží, ale jeho kvantitativní detekce se může podařit později v průběhu nemoci. Proto se má u subkorneální pustulózní dermatózy opakovat pátrání pro monoklonální gamapatii [21,52,53].
Histologické vyšetření kůže v případě klasického IgA pemfigu podobného subkorneální pustulózní dermatóze popisuje infiltraci polymorfonukleárními granulocyty v epidermis, převážně neutrofily s malým množstvím eozinofilů s formováním pustul a bul v různých vrstvách, dominantně však subkorneálně. Akantolýza není prominentní, může být přítomna ve starších ložiscích. Přímá a nepřímá imunofluorescence může být zpočátku negativní, teprve později se objeví detekovatelná ložiska IgA v epidermis (IgA pemfigus nebo IgA intercelulární dermatóza) [17].
Terminologicky je v této oblasti variabilita, která souvisí se vzácností této nemoci a rozdílností antigenů, na něž se váže IgA imunoglobulin.
Z léčebných alternativ se v literatuře uvádí dapson, ale zároveň se připouští, že odpovědi na tuto léčbu nebývají vždy kompletní [40]. Další léčebnou alternativou je acitretin a PUVA [41,42]. Tyto dva léčebné postupy jsme v našem případě nepoužili, protože byly popsány v případech nesouvisejících s monoklonální gamapatií.
Pulzní dávky dexametazonu a cyklofosfamidu se uvádějí jako účinná možnost léčby IgA pemfigu podobného subkorneální pustulózní dermatóze provázející monoklonální gamapatii [54].
Podobně jako u všech autoimunitních chorob, byly i u tohoto kožního onemocnění popsány léčebné odpovědi po aplikaci monoklonální protilátky antiCD20 s názvem rituximab (Mabthera) [57–63]. Nicméně žádný z popsaných léčebných postupů nezaručuje stoprocentní úspěch.
IgA pemfigus podobný subkorneální pustulózní dermatóze byl prvním a nadlouho jediným projevem monoklonální gamapatie. Teprve v rámci vyšetřování vezikulobulózního onemocnění se pacientka dostala na imunologii, kde zjistili monoklonální imunoglobulin, a tak byla v roce 2001 uzavřena hematologická diagnóza jako MGUS (Monoclonal Gammopathy of Undetermined Significance – monoklonální gamapatie nejistého významu).
Mimo symptomatické dermatologické léčby jsme pacientce po domluvě s dermatologem podali kombinovanou imunosupresivní léčbu, cyklofosfamid a dexametazon, a sledovali její vliv na kožní projevy a monoklonální imunoglobulin. Intenzita výsevů vezikulobulózních morf se při této léčbě zmenšila, ale kožní projevy zcela nevymizely, kožní léčebnou odpověď jsme hodnotili jako parciální remisi. Monoklonální imunoglobulin poklesl na nekvantifikovatelné množství, ale jeho stopy byly stále prokazatelné imunofixační elektroforézou (very good partial remission – VGPR). Zajímavé je, že zatímco nízké hladiny monoklonálního imunoglobulinu přetrvávaly alespoň 1/2 roku od ukončení léčby, intenzita vezikulobulózního postižení kůže se po ukončení léčby časně vrátila do původní podoby.
Na základě četných pozitivních zpráv z literatury o příznivém působení monoklonální antiCD20 – rituximabu (Mabthera) jsme se pokusili zmenšit míru utrpení 6 infuzemi 600mg rituximabu, které jsme podávali v týdenních intervalech. Efekt rituximabu byl však pouze dočasný, očekávaný léčebný úspěch se nedostavil.
Progresi monoklonální gamapatie do myelomu jsme jednoznačně prokázali koncem roku 2007, tedy po 6 letech sledování pro monoklonální gamapatii nejistého významu. Pro iniciální léčbu jsme zvolili standardní léčbu mnohočetného myelomu (cyklofosfamid, dexametazon a adriamycin). Po aplikaci 4 cyklů této léčby však nebyla zřetelná žádná hematologická léčebná odpověď.
Pro druhou léčebnou linii jsme zvolili thalidomid, dexametazon a cyklofosfamid. Tato léčba však jenom zhoršila kožní projevy až do obrazu erytrodermie s nutností hospitalizace. Thalidomid může vyvolávat makulózní kožní projevy a zde se zřejmě zkombinovaly nežádoucí kožní projevy thalidomidu s projevy základní nemoci. Zřejmě podávání thalidomidu pacientům s preexistujícím kožním onemocněním je spojeno se zvýšeným rizikem výskytu nežádoucích účinků thalidomidu. Kožní změny pacientů léčených thalidomidovými režimy bývají někdy způsobeny cotrimoxazolem, který je podáván v profylaktické dávce proti pneumocystě. V tomto případě však pacientka znovu měla profylaktické dávky cotrimoxazolu při kombinaci VCD, z čehož usuzujeme, že výrazné zhoršení kožních projevů nesouviselo v tomto případě s cotrimoxazolem, ale opravdu s thalidomidem.
Překvapivý úspěch však měla léčba třetí linie, bortezomib, cyklofosfamid a dexametazon.
Již po prvním měsíci této léčby klesl monoklonální imunoglobulin pod citlivost kvantitativní detekce a po 5 cyklech (měsících) léčby byla negativní i imunofixační elektroforéza. Již od druhého měsíce léčby se neobjevil nový výsev vezikulobulózních morf.
Závěr
Klinický průběh potvrzuje kauzální souvislosti IgA pemfigu podobného subkorneální pustulózní dermatóze s monoklonální gamapatií nejistého významu, která se transformovala v mnohočetný myelom.
Léčebná kombinace obsahující bortezomib navodila kompletní kožní i hematologickou remisi nemoci.
Léčebné schéma obsahující thalidomid zhoršilo kožní projevy nemoci až do obrazu erytrodermie. Rituximab byl v této indikaci neúčinný.
Tato práce vznikla a byla podporována v rámci projektu MŠMT: LC 06027 a VZ 0021622434.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e mail: z.adam@fnbrno.cz
Zdroje
1. Tichý M. Laboratorní analýza monoklonálních imunoglobulinů (paraproteinů). Český Těšín: Finidr 1997.
2. Tichý M. Paraproteinemie nejasného původu. Klin Biochem Metabol 1998; 6 : 172 – 175.
3. Ščudla V, Bačovský J, Indrák K et al. Výsledky léčby a změny prognózy nemocných s mnohočetným myelomem v období předchozích 40 let v oblasti střední a severní Moravy. Rozbor 562 nemocných. Vnitř Lék 2002; 48 : 707 – 717.
4. Špička I, Hájek R, Vytřasová M et al. Inhibitor proteazomu – bortezomib (Velcade) – v léčbě refrakterního mnohočetného myelomu. První zkušenosti v České republice. Čas Lék Čes 2005; 144 : 636 – 640.
5. Tichý M, Maisnar V. Laboratorní průkaz monoklonálních imunoglobulinů. Vnitř Lék 2006; 52 (Suppl 2): 41 – 45.
6. Tichý M, Řeháček V, Maisnar V et al. Monoklonální gamapatie v souboru 1 683 dárců plazmy. Čas Lék Čes 2004; 143 : 401 – 404.
7. Schnitzler L, Verret JL, Schubert B et al. Subcorneal pustulosis (Sneddon - Wilkinson disease) with acantholysis and IgA myeloma (13‑year follow‑up). Ann Dermatol Venereol 1977; 104 : 170 – 172.
8. Varigos GA. Subcorneal pustulosis with IgA abnormalities in serum and small bowel mucosa: case report. Australas J Dermatol 1979; 20 : 75 – 77.
9. Wallach E. Intraepidermal IgA pustulosis. J Am Acad Dermatol 1992; 27 : 993 – 1000.
10. Hodak E, David M, Ingber A et al. The clinical and histopathological spectrum of IgA pemphigus report of 2 cases. Clin Exp Dermatol 1990; 15 : 433 – 437.
11. Teraki Y, Amagai N, Hashimoto T et al. Intercellular IgA dermatosis of a childhood. Selective deposition of monomer IgA1 in the intercellular space of the epidermis. Art Dermatol 1991; 127 : 221 – 224.
12. Iwatsuki K, Hashimoto T, Ebihara T et al. Intercellular IgA vesiculo pustular dermatosis and related disorders: diversity of anti of anti IgA antiintecellular antoantibodies. Eur J Dermatol 1993; 3 : 7 – 11.
13. Cheng S, Edmonds E, Ben ‑ Gashir M et al. Subcorneal pustular dermatosis: 50 years on. Clin Exp Dermatol 2008; 33 : 229 – 233.
14. Ongenae KC, Temmerman LJ, Vermander F et al. Intercellular IgA dermatosis. Eur J Dermatol 1999; 9 : 85 – 94.
15. Harati A, Brockmeyer NH, Altmeyer Pet al. Skin disorders in association with monoclonal gammopathies. Eur J Med Res 2005; 10 : 93 – 104.
16. Birnkrant MJ, Papadopoulos AJ, Schwartz RA et al. Pyoderma gangrenosum, acne conglobata and IgA gammopathy. Int J Dermatol 2003; 42 : 213 – 216.
17. Rongioletti F, Patterson JW, Rebora A.The histological and pathogenetic spectrum of cutaneous diseases in monoclonal gammopathies. J Cutan Pathol 2008; 35 : 705 – 721.
18. Aste N, Fumo G, Pinna AL et al. IgA pemphigus of the subcorneal pustular dermatosis type associated with monoclonal IgA gammopathy. J Eur Acad Dermatol Venereol 2003; 17 : 725 – 727.
19. Sriprakash K, Yong‑Gee S. Multiple minute digitate hyperkeratoses associated with paraproteinaemia. Australas J Dermatol 2008; 49 : 233 – 236.
20. Bernard P, Amici JM, Bedane C et al. Intra ‑ epidermal neutrophilic IgA dermatosis associated with IgA myeloma. Ann Dermatol Venereol 1990; 117 : 890 – 892.
21. Bölcskei L, Husz S, Hunyadi J et al. Subcorneal pustular dermatosis and IgA multiple myeloma. J Dermatol 1992; 19 : 626 – 628.
22. Bowden JR, Scully C, Eveson JW et al. Multiple myeloma and bullous lichenoid lesions: an unusual association. Oral Surg Oral Med Oral Pathol 1990; 70 : 587 – 589.
23. Miyagawa S, Hashimoto T, Ohno H et al. Atypical pemphigus associated with monoclonal IgA gammopathy. J Am Acad Dermatol 1995; 32 : 352 – 357.
24. Niimi Y, Kawana S, Kusunoki T. IgA pemhigus. A case report and its characteristics clinical features compared with subcorneal pustular dermatosis. J Am Acad Dermatol 2000; 43 : 546 – 549.
25. Guana AL, Cohen PR. Transient acantholytic dermatosis in oncology patients. J Clin Oncol 1994; 12 : 1703 – 1709.
26. Ahmad K, Ramsay B. Pyoderma gangrenosum associated with subcorneal pustular dermatosis and IgA myeloma. Clin Exp Dermatol 2009; 34 : 46 – 48.
27. Andersen BL. Pyoderma gangrenosum associated with transient acantholytic dermatosis (pemphigus erythematosus‑like) and paraproteinemia. Acta Derm Venereol 1981; 61 : 77 – 79.
28. Horton JJ, Trounce JR, MacDonald DM. Bullous pyoderma gangrenosum and multiple myeloma. Br J Dermatol 1984; 111 : 227 – 230.
29. Ray R, Kanwar AJ, Ravichandran P et al. Pemphigus vulgaris with cryoglobulinemia and cold agglutinin disease. J Assoc Physicians India 1994; 42 : 420 – 422.
30. Kurokawa M, Koketsu H, Oda Y et al. A case of pemphigus vulgaris accompanied by multiple myeloma. Int J Dermatol 2005; 44 : 873 – 875.
31. Zhu X, Zhang B. Paraneoplastic pemphigus. J Dermatol 2007; 34 : 503 – 511.
32. Stiefelhagen P. Recurrent blood blisters on hands and face. The skin is a mirror of the bone marrow. MMW Fortschr Med 2007; 149 : 19.
33. Stone MS, Lyckholm LJ. Pyoderma gangrenosum and subcorneal pustular dermatosis. Clues to underlying imunoglobulin a myeloma. Am J Med 1996; 100 : 663 – 664.
34. Stoopler ET, Alawi F, Laudenbach JM et al. Bullous amyloidosis of the oral cavity: a rare clinical presentation and review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006; 101 : 734 – 740.
35. Lautenschlager S, Itin PH, Hirsbrunner Pet al. Subcorneal pustular dermatosis at the injection site of recombinant human granulocyte ‑ macrophage colony ‑ stimulating factor in a patient with IgA myeloma. J Am Acad Dermatol 1994; 30 : 787 – 789.
36. Kirsner RS, Anhalt GJ, Kerdel FA. Treatment with alpha interferon associated with the development of paraneoplastic pemphigus. Br J Dermatol 1995; 132 : 474 – 478.
37. Dal Tio R, Di Vito F, Salvi F. Subcorneal pustular dermatosis and IgA myeloma. Dermatologica 1985; 170 : 240 – 243.
38. Engineer L, Dow EC, Braverman IM et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol 2002; 47 : 943 – 946.
39. Lutz ME, Daoud MS, McEvoy MT et al. Subcorneal pustular dermatosis: a clinical study of ten patients. Cutis 1998; 61 : 203 – 208.
40. Roger H, Thevenet JP, Souteyrand P. Subcorneal pustular dermatosis associated with rheumatoid arthritis and raised IgA. Simultaneous remission of skin and joint involvement with dapsone treatment. Ann Rheum Dis 1990; 49 : 190 – 191.
41. Todd DJ, Bingham EA, Walsh M et al. Subcorneal pustular dermatosis and IgA paraproteinemia: response to etretinate and PUVA. Br J Hematol 1991; 125 : 387 – 389.
42. Takata M, Inaoki M, Shodo M et al. Subcorneal pustular dermatosis associated with IgA myeloma and intraepidermal IgA deposits. Dermatology 1994; 189 (Suppl 1): 111 – 114.
43. Atukorala DN, Joshi RK, Abanmi A et al. Subcorneal pustular dermatosis and IgA myeloma. Dermatology 1993; 187 : 124 – 126.
44. Vaccaro M, Cannavò SP, Guarneri B. Subcorneal pustular dermatosis and IgA lambda myeloma: a uncommon association but probably not coincidental. Eur J Dermatol 1999; 9 : 644 – 646.
45. Kasha EE jr, Epinette WW. Subcorneal puslutal dermatosis (Sneddon Wilkinson disease in association with monoclonal IgA gammopathy. Report and review of the literature. J Am Acad Dermatol 1988; 19 : 854 – 858.
46. Perera GK, Devereux S, Mufti G et al. PNP with Waldenström’s macroglobulinaemia. Clin Exp Dermatol 2005; 30 : 27 – 29.
47. Petropoulou H, Politis G, Panagakis P et al. Immunoglobulin A pemphigus associated with immunoglobulin A gammopathy and lung cancer. J Dermatol 2008; 35 : 341 – 345.
48. Vincendeau P, Claudy A, Thivolet J et al. Bullous dermatosis and myeloma. Monoclonal anticytoplasmic antibody activity. Arch Dermatol 1980; 116 : 681 – 682.
49. Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol 2008; 33 : 94 – 96.
50. Wong DA, Hunt MJ, Stapleton K. IgA multiple myeloma presenting as an acquired bullous disorder. Australas J Dermatol 1999; 40 : 31 – 34.
51. Ochiai T, Morishima T, Hao T et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol 2001; 28 : 407 – 411.
52. Ohno H, Miyagawa S, Hashimoto T et al. Atypical pemphigus with concomitant IgG and IgA anti‑intercellular autoantibodies associated with monoclonal IgA gammopathy. Dermatology 1994; 189 (Suppl 1):115 – 116.
53. Radfar L, Fatahzadeh M, Shahamat Y et al Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist 2006; 26 : 159 – 163.
54. Becker LR, Bastian BC, Wesselmann Uet al. Paraneoplastic pemphigus treated with dexamethasone/ cyclophosphamide pulse therapy. Eur J Dermatol 1998; 8 : 551 – 553.
55. Meijs M, Mekkes J, van Noesel C et al. Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma without Castleman’s disease; treatment with rituximab. Int J Dermatol 2008; 47 : 632 – 634.
56. Schadlow MB, Anhalt GJ, Sinha AA. Using rituximab (anti‑CD20 antibody) in a patient with paraneoplastic pemphigus. J Drugs Dermatol 2003; 2 : 564 – 567.
57. Fernando SL, O‘Connor KS. Treatment of severe pemphigus foliaceus with rituximab. Med J Aust 2008; 189 : 289 – 290.
58. Jessop S, Khumalo NP. Pemphigus: a treatment update. Am J Clin Dermatol 2008; 9 : 147 – 154.
59. Schierl M, Foedinger D, Geissler K et al. Paraneoplastic pemphigus despite treatment with rituximab, fludarabine and cyclophosphamide in chronic lymphocytic leukemia. Eur J Dermatol 2008; 18 : 717 – 718.
60. Schmidt E, Bröcker EB, Goebeler M. Rituximab in treatment‑resistant autoimmune blistering skin disorders. Clin Rev Allergy Immunol 2008; 34 : 56 – 64.
61. Sorce M, Aricò M, Bongiorno MR. Rituximab in refractory pemphigus vulgaris. Dermatol Ther 2008; 21 (Suppl 1): S6 – S9.
62. Zambruno G, Borradori L. Rituximab immunotherapy in pemphigus: therapeutic effects beyond B ‑ cell depletion. J Invest Dermatol 2008; 128 : 2745 – 2747.
63. Bölcskei L, Husz S, Hunyadi J et al. Subcorneal pustular dermatosis and IgA multiple myeloma. J Dermatol 1992; 19 : 626 – 628.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2009 Číslo 10
- Jak zlepšit záchyt a péči o osoby s prediabetem v primární péči?
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
- Hydroresponzivní krytí v epitelizační fázi hojení rány
- Význam hydratace při hojení ran
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
Najčítanejšie v tomto čísle
- Refluxní choroba jícnu. Standardy České gastroenterologické společnosti – aktualizace 2009
- Léčba refluxní choroby jícnu – současný stav
- Polohový test – víme o něm opravdu všechno?
- Neuroendokrinní nádory žaludku