
Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis
The Epstein-Barr nuclear antigen 3C (EBNA3C), one of the essential latent antigens for Epstein-Barr virus (EBV)-induced immortalization of primary human B lymphocytes in vitro, has been implicated in regulating cell proliferation and anti-apoptosis via interaction with several cellular and viral factors. Gemin3 (also named DDX20 or DP103) is a member of DEAD RNA helicase family which exhibits diverse cellular functions including DNA transcription, recombination and repair, and RNA metabolism. Gemin3 was initially identified as a binding partner to EBNA2 and EBNA3C. However, the mechanism by which EBNA3C regulates Gemin3 function remains unclear. Here, we report that EBNA3C directly interacts with Gemin3 through its C-terminal domains. This interaction results in increased stability of Gemin3 and its accumulation in both B lymphoma cells and EBV transformed lymphoblastoid cell lines (LCLs). Moreover, EBNA3C promotes formation of a complex with p53 and Gemin3 which blocks the DNA-binding affinity of p53. Small hairpin RNA based knockdown of Gemin3 in B lymphoma or LCL cells remarkably attenuates the ability of EBNA3C to inhibit the transcription activity of p53 on its downstream genes p21 and Bax, as well as apoptosis. These findings provide the first evidence that Gemin3 may be a common target of oncogenic viruses for driving cell proliferation and anti-apoptotic activities.
Published in the journal:
Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002418
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002418
Summary
The Epstein-Barr nuclear antigen 3C (EBNA3C), one of the essential latent antigens for Epstein-Barr virus (EBV)-induced immortalization of primary human B lymphocytes in vitro, has been implicated in regulating cell proliferation and anti-apoptosis via interaction with several cellular and viral factors. Gemin3 (also named DDX20 or DP103) is a member of DEAD RNA helicase family which exhibits diverse cellular functions including DNA transcription, recombination and repair, and RNA metabolism. Gemin3 was initially identified as a binding partner to EBNA2 and EBNA3C. However, the mechanism by which EBNA3C regulates Gemin3 function remains unclear. Here, we report that EBNA3C directly interacts with Gemin3 through its C-terminal domains. This interaction results in increased stability of Gemin3 and its accumulation in both B lymphoma cells and EBV transformed lymphoblastoid cell lines (LCLs). Moreover, EBNA3C promotes formation of a complex with p53 and Gemin3 which blocks the DNA-binding affinity of p53. Small hairpin RNA based knockdown of Gemin3 in B lymphoma or LCL cells remarkably attenuates the ability of EBNA3C to inhibit the transcription activity of p53 on its downstream genes p21 and Bax, as well as apoptosis. These findings provide the first evidence that Gemin3 may be a common target of oncogenic viruses for driving cell proliferation and anti-apoptotic activities.
Introduction
Epstein-Barr virus (EBV) is the first identified human tumor virus which causes infectious mononucleosis [1],[2], and is linked several lymphoproliferative diseases [3], including Burkitt’s lymphoma [4], nasopharyngeal carcinoma [5], and Hodgkin’s disease [6]. EBV, a ubiquitous human γ-herpesvirus, infects more than 90% of the worldwide adult population. Moreover, AIDS patients or post-transplant patients whose immune system is suppressed have a high probability of developing EBV-associated lymphomas. In vitro, EBV can transform normal resting human B-cells to continuous proliferation of latently infected B cells resulting in lymphoblastoid cell lines (LCLs). Like other herpesvirus, the EBV life cycle exhibits both latent (non-productive) and lytic (productive) phases. Four types of latency programs are classified: Type I latency is mainly represented by Burkitt lymphoma (BL) cells; there are two EBER genes expressed, the BART transcripts, and EBNA1 (EBV nuclear antigen 1) [7]. Type II latency is most frequently seen in Hodgkin's lymphoma and nasopharyngeal carcinoma with three additional latent-membrane proteins, LMP-1, LMP-2A and LMP-2B expressed [8]. Latency III is seen predominantly in lymphoproliferative diseases developed in immunocompromised individuals and EBV-transformed lymphoblastoid cell lines [3] . In this group all six EBNAs, three LMPs and the two EBERs are expressed [7],[9]. Type IV latency is less strictly defined and is associated with infectious mononucleosis patients [8],[9]. Some infected individuals is associated with the tightly latent program (latency 0), in which latent gene expression is almost undetectable [9]. The essential mediators for EBV transformation of B lymphocytes and establishment of latency in vitro include EBNA2, EBNA3A, 3C and LMP1 proteins. Importantly, EBNA3C plays a complex regulatory role in the transcription of several viral and cellular genes. For example, EBNA3C targets the cellular transcription factor RBP-Jκ to antagonize EBNA2 mediated transactivation [10],[11], and cooperates with EBNA2 to activate the major viral LMP1 promoter via interaction with the cellular transcription factor, Spi-1/Spi-B [12]. EBNA3C also regulates chromatin remodeling by recruiting both histone acetylase and deacetylase activities [13]–[15]. Furthermore, EBNA3C modulates the transcription of cellular genes involved in cell migration and invasion by targeting the metastasis suppressor Nm23-H1 [16]. In addition to its transcriptional functions, it has been reported that EBNA3C has cell-cycle regulatory functions, presumably mediated by direct protein–protein interactions [13],[17]–[19]. EBNA3C also stabilizes c-Myc and interacts with Mdm2 in modulating p53 transcription and apoptotic activities [20]–[22].
Gemin3 was discovered as a binding partner with EBV latent antigens (EBNA2 and EBNA3C), and the survival motor neuron (SMN) protein [23],[24]. It consists of 825 amino acids with 9 conserved motifs including the ASP-Glu-Ala-Asp motifs (or DEAD box motif), and belongs to DExD/H RNA helicase family, which plays many roles in RNA metabolism including pre-mRNA splicing, ribosome biogenesis, RNA transport, translation initiation, and RNA decay [24]–[28]. Both EBNA2 and EBNA3C combine with Gemin3 and SMN,while EBNA2 cooperate with SMN in transactivation of the viral LMP1 promoter [24]–[26]. It has been shown that Gemin3 also interacts with and modulates a variety of cellular transcription factors including steroidogenic factor 1(SF-1) [29],[30], early growth response protein 2 (Egr2) [31], forkhead transcription factor FOXL2 [32] , and mitogen Ets repressor METS [33]. Although Gemin3 was shown to play a role in gene transcription regulation, the function of Gemin3 in cell proliferation remains largely unclear. Here we show that Gemin3 is stabilized by EBNA3C via protein-protein interaction and can play a role in cell proliferation through formation of a complex with p53 which results in blocking p53-mediated transcriptional activity and apoptosis pathway. Knockdown of Gemin3 by RNA interference dramatically increases apoptosis of EBV-infected LCLs.
Results
EBNA3C forms a complex and co-localizes with Gemin3 in vivo
Similar to a screen by Grundoff, et al [24], a previous yeast two-hybrid study showed that C-terminal domain of EBNA3C interacts with Gemin3. To confirm that these two molecules do associate in vivo, we performed co-immunoprecipitation assays to determine if EBNA3C forms a complex with Gemin3 in EBV transformed B lymphoma cell lines (LCL1 and LCL2). The B lymphoma cell line Ramos which lacks EBNA3C expression because it is EBV negative was used as a control. The results showed that endogenous Gemin3 was dramatically immunoprecipitated by anti-EBNA3C antibody, but not by control mouse serum (Figure 1A). The reverse immunoprecipitation with anti-Gemin3 antibody further confirmed that EBNA3C does form a complex with Gemin3 in vivo (Figure 1B). Since Gemin3 is initially identified as a binding protein of EBNA2 and EBNA3C, we wanted to determine if the EBNA3C complex with Gemin3 was independent of EBNA2 and performed similar coimmunoprecipitation assays again by using the EBV negative B lymphoma BJAB derived cell lines BJAB7 and BJAB10 with EBNA3C stable expression alone. The results showed that EBNA3C alone was sufficient to form a complex with Gemin3 in vivo (Figure 1C).

To determine if the association of EBNA3C and Gemin3 occurs in the same cellular compartments in this scenario, we performed immunofluorescence assays by ectopically expressing FLAG-tagged EBNA3C and GFP-tagged Gemin3 in U2OS cells. The immunofluorescence results showed that although EBNA3C signals are shown as stippled, punctate dots with the exclusion of nuclei, and Gemin3 signal was displayed as intense staining of prominent but discrete foci in both nucleus and cytoplasm, and there were a number of positions where co-localization staining of EBNA3C with Gemin3 was evident (Figure 2A). To further visualize the interaction of these two proteins under physiological conditions, an EBV transformed cell line LCL1 was used. Endogenous EBNA3C and Gemin3 signals were evident with the co-localization pattern (Figure 2B), supporting the above data that these two proteins can associate in the same complex.

EBNA3C interacts with Gemin3 through its C-terminal domains
To define which domain of EBNA3C interacts with Gemin3, we co-transfected HEK293 cells with expression constructs for GFP-tagged Gemin3 and FLAG-tagged full-length (residues 1 to 992) or a series of truncated mutant of EBNA3C (1–365, 366–620, or 621–992), and performed coimmunoprecipitation assays with GFP or FLAG antibodies. Consistently, the results showed that coimmunoprecipitation targeting Gemin3 or EBNA3C, always results in Gemin3 co-immunoprecipitating with both the full-length and C-terminal domain of EBNA3C with relatively high affinity (Figure 3A and B). To further determine if the C-terminal domain of EBNA3C alone is sufficient to bind with Gemin3, we performed GST pulldown assays by in vitro-translating the full length and truncated mutants of EBNA3C followed by co-incubation with bacterially expressed GST-Gemin3 protein. The results showed that the C-terminal domain (621–992) of EBNA3C directly interacted with Gemin3 (Figure 3C). Similarly, using in vitro-translated Gemin3 coincubated with different bacterially expressed GST-EBNA3C (1–365, 366–581, 582–792, and 900–992), we further found that the C-terminal amino acids 621 to 792, a smaller region than previously identified by yeast two-hybrid analysis (534–778aa, [24]) of EBNA3C was critical for binding to Gemin3 in vitro (Figure 3D and F). To determine the domain of Gemin3 responsible for interacting with EBNA3C, we generated three truncated mutants of GST-Gemin3 (residues 1–272, 307–547 and 546–825) and performed GST pulldown assays with in vitro-translated full-length EBNA3C. The result showed that Gemin3 bound with EBNA3C via its C-terminal domain (Figure 3E and F), supporting the previously identified data by yeast two-hybrid analysis [24], and further narrows the interacting domain.
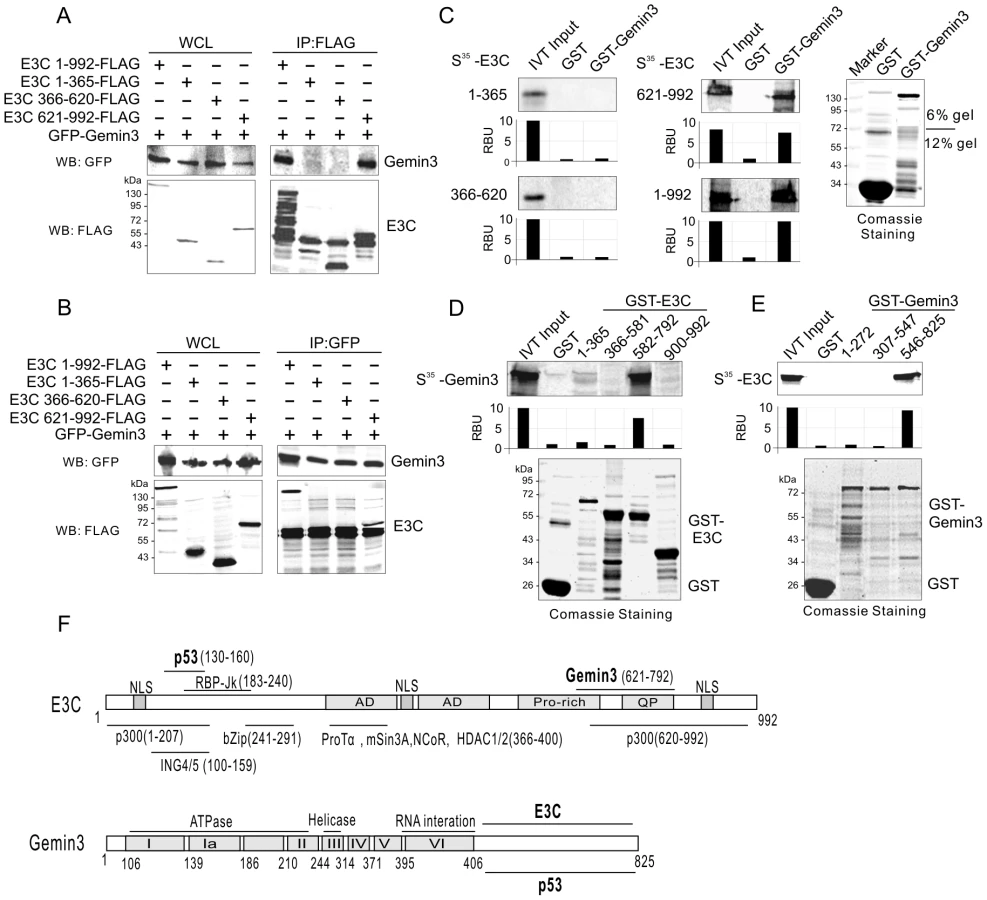
EBNA3C enhances the protein stability and production of Gemin3
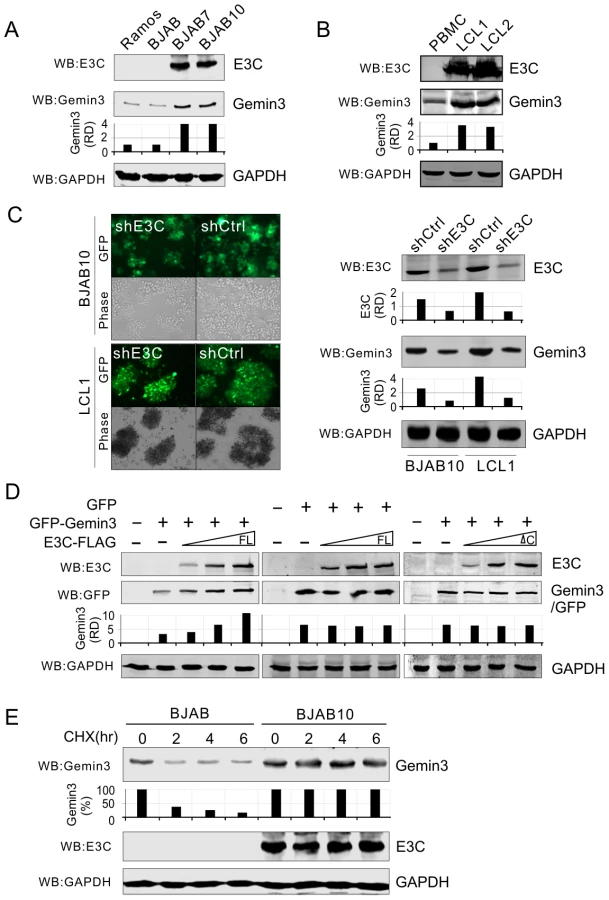
To determine if EBNA3C plays a role on the production of Gemin3, we tested endogenous Gemin3 protein levels in EBNA3C stable expressing cell lines (BJAB7 and BJAB10) and parental BJAB cells, as well as EBV negative Ramos cells. Interestingly, the results showed that EBNA3C greatly induced the production of Gemin3 in BJAB cells (Figure 4A). To verify if whole EBV latent infection with EBNA3C expression also has the same effect on Gemin3 production, we did a western blot to detect Gemin3 protein levels in both PBMC and two EBV-infected derived LCL cell lines. Consistent with EBNA3C expression alone, the results showed that Gemin3 protein levels were significantly increased by EBV infection (Figure 4B). The results of Gemin3 protein levels in EBNA3C-knockdown BJAB10 and LCL cells were consistently much lower than in the control cells (Figure 4C), further suggesting that EBNA3C is required for maintenance of Gemin3 levels. To determine if the increased production of Gemin3 is due to post-translational regulation by EBNA3C, we co-expressed exogenous EBNA3C-FLAG with GFP-Gemin3 or GFP control vector into Saos-2 cells and determined the protein levels of GFP-Gemin3 and GFP. The results showed that the production of GFP-Gemin3 but not GFP was enhanced by EBNA3C in a dose-dependent manner (Figure 4D, left and middle panels), indicating that the interaction of EBNA3C with Gemin3 is important for Gemin3 accumulation. To this end, we carried out similar experiments by using an EBNA3C mutant lacking Gemin3-interacting domain, and found that deletion of the Gemin3-interacting domain efficiently abolished the effect of EBNA3C on Gemin3 production (Figure 4D, left and right panels). To further confirm that EBNA3C-induced Gemin3 levels were due to enhanced Gemin3 protein stability, we performed the protein stability assays for Gemin3 by treating BJAB (EBNA3C negative) and BJAB10 (EBNA3C positive) cells with cycloheximide (a protein translation inhibitor). The results showed that the stability of Gemin3 was dramatically enhanced in the presence of EBNA3C compared to cells without EBNA3C (Figure 4E).
Gemin3 interacts with p53 and contributes to EBNA3C-mediated inhibition of p53 transcriptional activity
Our previous studies showed that EBNA3C blocks p53-mediated transcriptional as well as apoptotic activity [22],[34]. Gemin3 plays an essential role in cell survival and can function as a transcriptional repressor through interaction with a number of transcriptional factors [35],[36]. Therefore, we hypothesized that EBNA3C enhanced Gemin3 levels may be due to its interaction with p53 and in turn repress the transcriptional activity of p53. To this end, we first asked if Gemin3 can directly bind to p53 in vitro. Using in vitro-translated Gemin3, we incubated it with bacterially expressed GST or GST-p53 protein. We found that Gemin3 strongly bound to p53 but not the GST control (Figure 5A, lane 2 and 3). Using different truncation mutants of p53 fusion with GST, we found that the DNA-binding domain (amino acids 100–300) of p53 is critical for interaction with Gemin3 (Figure 5A, lane 4 to 6). To define the binding site of Gemin3 with p53, we utilized full length and truncated mutants of Gemin3 fusion with GST to incubate with in vitro-translated p53. The results showed that the C-terminal domain (amino acids 546–825) alone of Gemin3 presents similar strong binding with p53 as full length Gemin3 (Figure 5B). This further confirms that Gemin3 can directly interact with p53. To test if endogenous p53 interacts with Gemin3 in cells and whether this interaction is impaired by EBNA3C, we performed immunoprecipitation assays with anti-p53 antibody followed by western blotting with Gemin3 antibody individually from cell lysates of BJAB, BJAB10 and LCL1 cells. The results showed that Gemin3 and p53 do form a complex in EBV negative BJAB cells, and that the association of p53 with Gemin3 was greatly enhanced by 2.2 fold in EBNA3C-expressing BJAB10 cells and 5.2 fold in LCL1 cells when compared to that in the EBV negative BJAB cells (Figure 5C, bottom panel). Since Gemin3 bound to the DNA-binding domain of p53, we wanted to know if Gemin3 blocks sequence specific DNA-binding ability of p53, and so performed chromatin immunoprecipitation assays with exogenous p53 alone or p53 and Gemin3 in the presence and absence of p53-responsive DNA element (p53-RE). As shown in the Figure 5D, Gemin3 dramatically reduced the ability of p53 to bind to the p53-responsive DNA element. To further confirm the consequence of Gemin3 blocking DNA-binding ability of p53 and argue the possibility that Gemin3 can directly bind to DNA probe, we also performed reporter assays by co-expressing exogenous HA-p53 with increasing doses of GFP-tagged Gemin3 in the presence of 13 copies of p53-binding sites driven luciferase reporter into p53-null Saos-2 cells. The results showed that Gemin3 diminished p53-mediated transcriptional activity in a dose-dependent manner (Figure 5E). The expression levels of Gemin3, p53, and GAPDH as a loading control, were also analyzed by western blotting (Figure 5E, bottom panels). To answer if EBNA3C-mediated inhibition of p53 transcriptional activity is dependent on Gemin3 accumulation, we performed similar reporter assays by co-expressing p53 with or without EBNA3C in the presence of specific Gemin3 shRNA (shG3) or non-specific control shRNA (shCtrl). Strikingly, the results showed that EBNA3C-mediated inhibition of p53 transcriptional activity is dramatically reversed once Gemin3 is knocked down (Figure 5F, compare lane 3 with 6). This suggests that Gemin3 is important for EBNA3C-mediated inhibition of p53 function.

Gemin3 knockdown attenuates EBNA3C-mediated inhibition of p53-induced apoptosis
Studies in our lab have shown that EBNA3C is able to inhibit p53-induced apoptosis in p53-null Saos2 cells [22],[34]. To further confirm the significance of Gemin3 on EBNA3C-mediated inhibition of p53 function, we verified whether or not Gemin3 knockdown affects EBNA3C-mediated inhibition of p53-induced apoptosis by using colony formation assays. The results showed that coexpression of EBNA3C with p53 markedly increased the colony formation of Saos-2 cell compared to that produced by p53 alone (Figure 6A, left panel). In contrast, although less colony formation was produced in the vector alone with the Gemin3 knockdown (shG3) compared to that with the control knockdown (shCtrl) (Figure 6A, top panel), the colony formation of p53 co-expressed with EBNA3C was significantly inhibited in Gemin3 knockdown cells (Figure 6A). Thus, these data revealed a role for EBNA3C in upregulation of Gemin3 for anti-apoptosis and promotes cell proliferation.

To further prove that Gemin3 plays an important role in cell proliferation, we performed apoptosis assays using B lymphoma cells or LCLs with or without lentivirus-mediated Gemin3 knockdown. Gemin3 expression was significantly knocked down in the EBV negative B lymphoma cell lines Ramos and BJAB, EBV transformed B cell line LCL1 as well as EBNA3C stably expressing BJAB cell lines BJAB7 and BJAB10 (Figure 6B). The results of apoptotic assays showed that Gemin3 knockdown cells had a dramatic increase in apoptosis compared to the control knockdown cells (Figure 6C). Furthermore, Gemin3 knockdown in EBV or EBNA3C positive cells showed significantly higher levels of apoptosis than that in EBV or EBNA3C negative cells (Figure 6C, bottom panel). Consistently, the quantitative PCR analysis showed that the transcriptional levels of p53 downstream genes including p21, Bax and p53 were enhanced when Gemin3 was knocked down in EBNA3C positive cells and were remarkably higher than that in EBNA3C negative cells (Figure 6D). Therefore, Gemin3 plays a critical role in cell proliferation in EBNA3C positive cells.
Discussion
Emerging evidence have indicated that Gemin3 is an essential gene for embryonic development and survival from mammal to Drosophila [23],[37]. Gemin3 (DP103/DDX20) was originally discovered in a screen for cellular factors that bind to EBNA2 and EBNA3C using the yeast two-hybrid system [24]. Gemin3 was shown to be involved in EBNA2-mediated transactivation and transformation, and a deletion mutant of EBNA2 lacking the Gemin3-binding site severely impeded LMP1 transactivation and viral transformation [26]. However, there was no previous report on the functional effect of EBNA3C on Gemin3. Our yeast two-hybrid screen showed that Gemin3 strongly interacted with EBNA3C and this was identified with high frequency in positive yeast clones when the C-terminal domain of EBNA3C was used as bait (data not shown). We now confirm that Gemin3 directly binds to EBNA3C in vitro and in vivo, and this interaction led to increased Gemin3 protein stability and its accumulation in B lymphoma cells and EBV-transformed lymphoblastoid cell lines. Furthermore, EBNA3C promoted the formation of a complex with p53 and Gemin3 in vivo which was important for blocking p53-mediated transcriptional activity and apoptosis. Inhibition of Gemin3 expression dramatically abolished EBNA3C-mediated inhibition of p53-induced apoptosis (Figure 7). The emerging evidence showing Gemin3 as a DEAD-box RNA helicase that plays a role in carcinogenesis, will inspire us to develop efficient helicase inhibitors as a potential target for anti-cancer in the future.

Gemin3 is also an integral component of the SMN complex, which plays an essential role in the production of spliceosomal small nuclear ribonucleoproteins (snRNPs) [38], regulation of DNA transcription [30], pre-mRNA splicing [37], and axonal RNA transport [39]. In addition to interaction with Gemin3, EBNA3C also has been shown to associate with SMN [25]. This further supports the notion that EBNA3C targets the SMN complex and can impair the function of each component including Gemin3 in the SMN complex. Here, we demonstrate a novel function of Gemin3 as a regulator that suppresses p53-mediated transcriptional activity and apoptosis. Interestingly, SMN was also shown to interact with p53 [40]. Therefore, our results showing that knockdown of Gemin3 sufficiently rescues p53, suggest that Gemin3 could be a critical member of the SMN complex to suppress p53 function. This provides an explanation as to why EBNA3C promotes complex formation of p53 and Gemin3 through enhancing Gemin3 protein stability.
The N-terminal domain of Gemin3 contains the conserved helicase motifs, while the non-conserved C-terminal domain can interact with a variety of cellular and viral factors. In this study, we found that Gemin3 binds with both EBNA3C and p53 through the same region (amino acid 548 to 825) of its C-terminal domain. Considering that EBNA3C individually binds to p53 and Gemin3 through its N-terminal and C-terminal domains. There is a strong possibility for EBNA3C to act as an adaptor to promote Gemin3 interaction with p53. This also may provide an explanation as to why the levels of p53 in complex with Gemin3 are dramatically enhanced in the presence of EBNA3C.
Although it has been shown that Gemin3 plays a diverse roles on repressing gene transcription [30], the mechanism of Gemin3-mediated transcriptional repression is not fully clear. One of the potential mechanisms could be through recruitment of specific transcription co-repressors. For examples, Gemin3 interacts with METS to repress Ets by assembling a complex of N-CoR, Sin3A, HDAC-2, and HDAC-5 [30],[33],[41]. Gemin3 was also found to form a complex with METS (mitogenic Ets transcriptional suppressor)/PE1 or ERF (Ets2 repressor factor) controlling cellular proliferation and differentiation through recruitment of HDAC2 and HDAC5 [42]. It also interacts with and represses the transcriptional activity of Egr2/Krox-20 which is dependent in part on HDAC recruitment [31]. Gemin3 also interacts with the transcriptional factor FOXL2 important for inducing apoptosis [32]. However, our study showed that Gemin3 reduces p53 transcriptional activity and now provides another potential mechanism through which Gemin3 can sequester a transactivation cofactor or may disrupt a transcriptionally active nucleic acid-protein complex.
Previous studies in our lab indicated that EBNA3C targets multiple factors to deregulate the p53 pathway and so drive cell proliferation. For example, EBNA3C has a unique activity of ubiquitylation and de-ubiquitylation [17],[18],[21], and also suppresses p53 function through formation of a complex with Mdm2 [21]. More recently, our lab also showed that EBNA3C directly competes with two p53-associated growth inhibitors ING4 and ING5 for attenuating p53 function [34]. Despite current studies which convincingly showed that EBNA3C inhibit p53 function, it is still unknown as to why the protein level of p53 is not affected in EBV-transformed LCLs or in B lymphoma expressing EBNA3C [43]–[46]. Here, our study further addresses a potential mechanism that EBNA3C can block p53 function through upregulating Gemin3 protein levels and so promoting the formation of a complex of p53 with Gemin3. This is distinctly different from the strategy used by KSHV encoded LANA where p53 ubiquitylation is induced and results in its degradation [47]. In summary, this study provides initial evidence that Gemin3 can directly interact with p53 through its DNA-binding domain and in turn inhibits its activities. This pathway is essentially targeted by EBNA3C in EBV-transformed lymphoblastoid cells.
Materials and Methods
DNA constructs and antibodies
pA3F-EBNA3C constructs expressing either full-length EBNA3C or different truncated versions of EBNA3C 1–365, 366–620, and 621–992 with a Flag tag at the carboxy-terminal end and Glutathione S-transferase (GST)-EBNA3C truncation mutation were described previously [20]. Constructs expressing green fluorescent protein (GFP)-tagged Gemin3 was prepared by cloning PCR-amplified fragments into pEGFP-C1 vector (BD Biosciences Clontech) at EcoRI and SalI restriction sites. The plasmid pGL3-p53-RE contains 13 copies of p53-binding site at the upstream of firefly luciferase gene was constructed by EcoRV insertion of p53-binding sequences into the pGL-3 luciferase reporter plasmid (Kindly provided by Wafik S. EI-Deiry at University of Pennsylvania, Philadelphia, Pennsylvania). pGEX-p53 expresses an N-terminal glutathione S-transferase (GST)-p53 fusion protein was derived from pGEX-2T (Amersham Pharmacia, Inc, Piscataway, NJ) by insertion of human p53 cDNA (A gift from Gary J Nabel, National Institutes of Health, Bethesda, MD) at the BamH1 and EcoR1 sites. pcDNA-HA-p53 was generated by cloning PCR-amplified p53 cDNA using pGEX-p53 as a template into the pcDNA3-HA vector at EcoR1 and Not1 sites. GST-p53 truncation mutants were constructed by insertion of PCR fragments into the pGEX-5x-1 backbone (gift from Shelley L Berger, The Wistar Institute, Philadelphia, PA). Plasmid expressing GST-Gemin3 was kindly provided by Dr. Gideon Dreyfuss (Howard Hughes Medical Institute Investigator at University of Pennsylvania Perelman School of Medicine) and described previously [23]. GST-Gemin3 truncation mutants GST-Gemin3 1–272 was constructed by insertion of PCR fragments into pGEX-2TK derivative vector at Kpn1 and EcoR1 sites, GST-Gemin3 307–547 and GST-Gemin3 546–825 into pGEX-2TK vector BamH1 and EcoR1 sites. All constructs and mutations were verified by DNA sequencing.
Mouse monoclonal anti-Gemin3 (12H12) was kindly provided by Dr. Gideon Dreyfuss (Howard Hughes Medical Institute Investigator at University of Pennsylvania School of Medicine). The monoclonal antibodies mouse anti-myc (9E10) and anti-EBNA3C (A10) were prepared from the respective hybridoma cultures. Mouse monoclonal antibody anti-FLAG epitope (M2) was purchased from Sigma-Aldrich Corp. (St. Louis, MO). Mouse monoclonal antibody reactive to p53 (DO-1) and GFP (F56-BA1) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell culture and transfection
Human embryonic kidney cells transformed with sheared adenovirus type 5 DNA HEK293 cell line and p53-null cell line SaoS-2 were obtained from Jon Aster (Brigham and Woman’s Hospital, Boston, MA) [48]. U2OS is also a human osteosarcoma cell line [49]. EBV negative Burkitt’s lymphoma cell lines BJAB and Ramos were provided by Elliott Kieff (Brigham and Woman’s Hospital, Boston, MA) [50]. LCL1 and LCL2 are in vitro-transformed EBV positive cell lines. BJAB cells expressing EBNA3C (BJAB7 and BJAB10) were generated previously by transfecting with pZipneo eukaryotic expression vector with EBNA3C cDNA followed by neomycin selection [51]. HEK 293, U2OS and Saos-2 cells were grown in Dulbecco's modified Eagle's medium (purchased from HyClone, Logan, UT) supplemented with 10% fetal bovine serum, 50 U/ml penicillin, 50 µg/ml streptomycin, and 2 mM L-glutamine. BJAB, Ramos and the EBV-positive cell lines were maintained in RPMI 1640 medium (HyClone, Logan, UT) supplemented as described for Dulbecco's modified Eagle's medium above. All cultures were incubated at 37°C in a humidified environment supplemented with 5% CO2. Cells were transfected by electroporation with a Bio-Rad Gene Pulser II electroporator. Briefly, 15×106 cells harvested in exponential phase were collected, washed in phosphate-buffered saline (PBS), and resuspended in 400 µl of the appropriate medium containing DNA for transfection [52]. Resuspended cells were transferred to a 0.4-cm-gap cuvette, and electroporation was performed at 975 µF, 210 V for HEK293 and Saos-2 cells. Transfected cells were transferred to a 100-mm petri dish containing 10 ml of complete medium and incubated at 37°C.
Immunoprecipitation and Western blotting
Transfected cells were harvested, washed with ice-cold PBS, and lysed in 0.5 ml ice-cold radioimmunoprecipitation (RIPA) buffer (1% Nonidet P-40 [NP-40], 10 mM Tris [pH 7.5], 2 mM EDTA, 150 mM NaCl), supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, 1 µg/ml pepstatin, and 1 µg/ml leupeptin). Cell debris was removed by centrifugation at 21,000×g (10 min and 4°C), and the supernatant was transferred to a fresh microcentrifuge tube. Lysates were then precleared by end-over-end rotation with normal mouse serum and 30 µl of a 1∶1 mixture of Protein A-Protein G-conjugated Sepharose beads (1 h, 4°C). Beads were spun out, and supernatant was transferred to a fresh microcentrifuge tube and approximately 5% of the lysate was saved for input control. The protein of interest was captured by rotating the remaining lysate with 1 µg of appropriate antibody overnight at 4°C. Immune complexes were captured with 30 µl of a 1∶1 mixture of Protein A and Protein G Sepharose beads, pelleted, and washed five times with ice-cold RIPA buffer. For Western blot assays, input lysates and immunoprecipitated (IP) complexes were boiled in Laemmli buffer [53], fractionated by SDS-PAGE, and transferred to a 0.45 µm nitrocellulose membrane. The membranes were then probed with appropriate antibodies followed by incubation with appropriate infrared-tagged secondary antibodies and viewed on an Odyssey imager (LiCor Inc., Lincoln, NE).
Purification of GST fusion proteins
Escherichia coli BL21 (DE3) cells were transformed with the plasmid constructs for each GST fusion protein. Single colonies were picked and grown overnight in 3 ml of Luria broth supplemented with 100 µg/ml ampicillin. One milliliter of the overnight culture was used to inoculate a 500 ml culture. The larger culture was incubated until the optical density at 600 nm was approximately 0.6, at which point it was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 12 h at 30°C . The bacteria were pelleted, washed once with STE buffer (100 mM NaCl, 10 mM Tris, and 1 mM EDTA, pH 7.5), resuspended in 3 ml NETN buffer (0.5% NP-40, 100 mM NaCl, 20 mM Tris, 1 mM EDTA, pH 8.0), supplemented with protease inhibitors, and incubated on ice for 15 min. A volume of 150 µl of 1 M dithiothreitol (DTT) and 1.8 ml of a 10% solution of Sarkosyl in STE buffer was added, and the suspension was sonicated (for 3 min on ice) to solubilize the proteins. The lysates were centrifuged (12,000×g, 10 min, 4°C) to separate the unsolubilized fraction. The clear supernatant was transferred to a fresh tube, to which 3 ml of 10% Triton X-100 in STE buffer and 200 µl of Glutathione-Sepharose beads were added. The tube was rotated overnight at 4°C, after which the purified protein bound to Glutathione was collected by centrifugation (2 min, 600×g, 4°C) and washed five times with NETN buffer supplemented with protease inhibitors. The level of purification was determined by SDS-PAGE, and purified proteins were stored at 4°C.
GST pull-down assays
For pull-down assays from cell lysates, lysates were prepared in RIPA buffer (0.5% NP-40, 10 mM Tris [pH 7.5], 2 mM EDTA, 150 mM NaCl, supplemented with protease inhibitors). Lysates were precleared and then rotated with either GST control or the appropriate GST fusion protein bound to Glutathione-Sepharose beads. For in vitro binding experiments, GST fusion proteins were incubated with 35S-labeled in vitro-translated protein in binding buffer (1×PBS, 0.1% NP-40, 0.5 mM DTT, 10% glycerol, supplemented with protease inhibitors). In vitro translation was performed with the T7-TNT Quick Coupled transcription-translation system (Promega Inc., Madison, WI) according to the manufacturer's instructions.
Immunofluorescence
To check the co-localization of ectopically expressed GFP-Gemin3 and Flag-EBNA3C in the cells, we used Lipofectamine 2000 (Invitrogen, Carlsbad, CA) to transfect U2OS cells with the plasmids then cultured on coverslips. At 36 h posttransfection, cells were fixed using 3% paraformaldelhyde with 0.1% Triton X-100 for 20 min at room temperature. We used LCL1 cells to examine the co-localization of endogenous Gemin3 with EBNA3C, appropriate LCL1 cells were added onto slides and fixed using the same method after culture 5 h. Fixed cells were washed with PBS and subsequently blocked in 1% BSA for 10 min. Gemin3 was detected using mouse monoclonal antibody (12H12). Endogenous EBNA3C was detected using EBNA3C-reactive rabbit polyclonal antibody (1∶150 dilution); Flag-tagged EBNA3C was detected using M2 antibody (1∶1,000 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Primary antibodies were incubated with the cells for 60 min at the room temperature. Cells were washed three times with PBS and exposed to secondary antibodies. Goat anti-mouse antibody conjugated to Alexa Fluor 594 to detect Gemin3 and goat anti-rabbit antibody conjugated to Alexa Fluor 488 to detect EBNA3C were respectively used for LCL1 cells, and Goat anti-mouse antibody conjugated to Alexa Fluor 594 to detect FLAG-EBNA3C was used for U2OS cells. Secondary antibodies were diluted in blocking buffer at 1∶1,000 and incubated for 1 h at RT, followed by three washes with blocking buffer. The last wash contained 4′, 6′-diamidino-2-phenylindole (DAPI; Promega, Madison, WI) to counterstain the nuclei. The slides were examined using Olympus confocal microscopy and the images were analyzed with a Fluoview FV300 (Olympus, Melville, NY) software.
Luciferase reporter assay
Twelve million Saos-2 cells were co-transfected by using a Bio-Rad electroporater (Bio-Rad Laboratories, Inc., Hercules, CA) with different combination of pGL3-p53-RE, pcDNA-HA-p53, pA3F-EBNA3C and GFP-Gemin3. At 24 h post-transfection, cells were harvested, washed in PBS, and lysed by cell lysis buffer (BioVision, Inc., Mountain View, CA). Forty microliters of cell lysate was used for the reporter assay, using an LMaxII384 luminometer (Molecular Devices, Inc., Sunnyvale, CA). A portion of the cell lysate was used for Western blotting. Transferred proteins were detected with Odyssey infrared scanning technology (LI-COR, Inc., Lincoln, NE), using Alexa Fluor 680 and Alexa Fluor 800 (Molecular Probes, Carlsbad, CA, and Rockland, Gilbertsville, PA, respectively). All the transfections were performed multiple times, and the results shown represent the means of the data from three independent experiments.
Lentiviral-mediated gene silencing
For the lentivirus-mediated knockdown of EBNA3C or Gemin3, the EBNA3C shRNA sequence (CCAUAUACCGCAAGGAAUA) or Gemin3 shRNA (ACUCCCCAGUGAGACCAUU) was respectively inserted into pGIPZ vector according to the manufacture’s instructions (Open Biosystem, Inc, Huntsville, AL), the vector expressing EBNA3C or Gemin3 small hairpin RNA is abbreviated as shE3C or shG3, respectively. A 21-mer oligonucleotide (UCUCGCUUGGGCGAGAGUAAG) that had no significant homology to any known human mRNA in the databases was cloned in the same vector and used as control. Control shRNA is hereinafter abbreviated as shCtrl.
Lentiviruses were produced by transient transfection into HEK 293T cells as previously described with the following modifications [54]. A total of 2×106 293T cells were seeded in 10-cm-diameter dishes in DMEM (HyClone, Logan, UT) supplemented with 10% FBS and cultured for 24 h prior to transfection. A total 20 µg of plasmid DNA was used for the transfection of each dish, including 1.5 µg of envelope plasmid pCMV-VSV-G (catalog no. 8454; Addgene, Inc., Cambridge, MA), 3 µg of packaging plasmid pRSV-REV (catalog no. 12251 Addgene, Inc., Cambridge, MA), 5 µg of packaging plasmid pMDLg/Prre (catalog no. 12251 Addgene, Inc., Cambridge, MA), and 10.5 µg of lentiviral vector plasmid. The precipitation was formed by adding the plasmids to a final volume of 438 µl of H20 and 62 µl of 2 M CaCl2, mixing well, adding 500 µl of 2×HEPES-buffered saline, and then incubating at room temperature for 30 min. Chloroquine was added to the 10 ml of plated media with a final concentration of 25 µM at 5 minutes prior to transfection. The medium was replaced after 12 h with DMEM supplemented with 10% FBS and 10 mM HEPES, and 10 mM sodium butyrate. The medium was replaced again 10 hours later using DMEM supplemented with 10% FBS and 10 mM HEPES. The conditioned medium was collected four times at 12 h interval, filtered through 0.45 µm pore-size cellulose acetate filters, and stored on ice. The virus was concentrated by spinning at 70,000×g for 2.5 h. The concentrated virus was resuspended in RPMI then used to infect 106 cells in the presence of 20 µM/ml Polybrene. After 72 h, add puromycin to final concentration of 2 µg/ml for selection. GFP immunofluorescence was assessed by using an Olympus IX71 microscope filtered with 560-nm excitation and 645-nm emission filters. Visible colonies were grown to 80% confluence in the presence of 2 µg/ml puromycin prior to western blot and apoptosis analysis.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) experiments were done essentially as previously described with some modifications [55],[56]. Saos-2 cells (10×106) transfected HA-p53 with or without GFP-Gemin3 in the presence of pGL-p53-RE or pGL3-basic vector alone were cross-linked with 1.1% (v/v) formaldehyde, 100 mM NaCl, 0.5 mM EGTA, and 50 mM Tris-HCl (pH 8.0) in growth medium at 37°C for 10 min, then at 4°C for 50 min. Formaldehyde was quenched by adding 0.05 vol 2.5 M glycine. Fixed cells were washed with PBS, incubated for 15 min in 15 ml of 10 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.5 mM EGTA, and 0.25% (v/v) Triton X-100, followed by 15 min in 15 ml of 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, and 200 mM NaCl, and finally sonicated in 1 ml of 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% (w/v) SDS plus 1 mM PMSF, 1 µg/ml aprotonin, leupettin, and pestatin to an average fragment size of 300–500 bp. 20% of solubilized chromatin extracts were saved for input followed with cross-link reverse step, and the remaining were clarified by centrifugation at 12,000 g, and diluted to 6 OD260 U/ml in IP buffer [140 mM NaCl, 1% (w/v) Triton X-100, 0.1% (w/v) sodium deoxycholate, 1 mM PMSF, 100 µg/ml salmon sperm DNA, and 100 µg/ml BSA]; preincubated for 1 h at 4°C with 10 µl/ml 50% (v/v) Protein A-agarose (Invitrogen Life Technologies, Camarillo, CA) with normal mouse/rabbit sera; reconstituted in PBS, and washed several times in IP buffer. Aliquots (600 µl) were incubated with 20 µg of each specific antibody for overnight at 4°C. Immune complexes were separated into bound and unbound complexes with protein A-agarose and cross-links were reversed by treatment at 65°C overnight. After treatment with RNase A and proteinase K, samples were extracted once with phenol/chloroform, and the DNA was precipitated with 2 volumes of ethanol. Precipitated DNA was pelleted, washed once with 70% ethanol, dried, and resuspended in 100 µl of water. The DNA was analyzed by quantitative PCR using Ampicilin primers (forward: 5’-CATCTTACGGATGGCATGAC-3’, reverse: 5’-CAACGATCAAGGCGAGTTAC-3’).
Colony formation assay
Ten million of Saos-2 cells were typically transfected using electroporation with different combinations of expression plasmids as shown in the text. Transfected cells were cultured in the selection medium (DMEM supplemented with 100 mg/ml G418). After 14 days, cells were fixed on the plates with formaldehyde and stained with 0.1% crystal violet. The amount of the colonies in each dish was scanned by Li-Cor Odyssey and counted. The data are presented as the average from two independent experiments.
Quantitative real-time PCR
Total RNA from cells was extracted using Trizol reagent and cDNA was made with a Superscript II reverse transcription kit (Invitrogen, Inc., Carlsbad, CA). The primers for real-time PCR were as followings: for p53 : 5′-CCT GAGGTTGGCTCTGACTGTA-3′(sense) and 5′-TCCGTCCCAGTAGATTACCAC - 3′ (antisense), yielding a 136-bp product; for p21 : 5′-GAGGGCAAGTACGAGTGG CAA-3′ (sense) and 5′ - CTGCGCATTGCTCCGCTAACC-3′ (antisense), yielding a 170-bp product; for Bax: 5′ - TGCTTCAGGGTTTCATCCAGGA-3′ (sense) and 5′ - ACGGCGGCAATCATCCTCTG-3′ (antisense), yielding a 172-bp product; and for GAPDH (glyceraldehyde-3-phosphate dehydrogenase): 5′-CTCCTCTGACTTCAAC AGCG-3′ (sense) and 5′-GCCAAATTCGTTGTCATACCAG-3′ (antisense), yielding a 112-bp product. The cDNA was amplified by using 10 µl of Master Mix from the DyNAmo SYBR green quantitative real-time PCR kit (MJ Research, Inc.), 1 µM of each primer, and 2 µl of the cDNA product in a 20 µl total volume. Thirty cycles of 1 min at 94°C, 30 s at 55°C, and 40 s at 72°C were followed by 10 min at 72°C in an MJ Research Opticon II thermocycler (MJ Research, Inc., Waltham, MA). A melting curve analysis was performed to verify the specificities of the amplified products. The values for the relative levels of change were calculated by the “delta delta threshold cycle” (ΔΔCT) method and each sample were tested in triplicates.
Apoptosis assay
The apoptotic cells of stable Gemin3 and control knock down cells were analyzed by propidium iodide (PI) flow cytometric assay, which is based on the principle that DNA fragmentation and the consequent loss of nuclear DNA content occurs at the late phase of apoptosis. Briefly, 106 cells with serum starvation treatment of 0.1% serum for 12 h were collected and fixed with 100% ethanol for 2 h at 4°C, washed with 1x phosphate-buffered saline (PBS), and stained with 50 µg/ml propidium iodide (Sigma, St. Louis, MO) and 1 µg/ml RNase for 1 hour in the dark at 4°C. The stained cells were subseqeutly analyzed using FACSCalibur cytometer (Becton Dickinson, San Jose, CA) and FlowJo Software (Tree Star, Ashland, OR).
Zdroje
1. BurkittD 1958 A sarcoma involving the jaws in African children. Br J Surg 46 218 223
2. EpsteinMAAchongBGBarrYM 1964 Virus Particles In Cultured Lymphoblasts From Burkitt's Lymphoma. Lancet 1 702 703
3. RickinsonABKieffE 2002 Epstein-Barr virus. FieldsBNKnipeDMHowleyPM Fields virology, 4th ed Vol 2, Philadelphia Lippincott Williams & Wilkins 2575 2627
4. HenleGHenleWDiehlV 1968 Relation of Burkitt's tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc Natl Acad Sci U S A 59 94 101
5. zur HausenHSchulte-HolthausenHKleinGHenleWHenleG 1970 EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228 1056 1058
6. JohanssonBKleinGHenleWHenleG 1970 Epstein-Barr virus (EBV)-associated antibody patterns in malignant lymphoma and leukemia. I. Hodgkin's disease. Int J Cancer 6 450 462
7. KuppersR 2003 B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol 3 801 812
8. Thorley-LawsonDA 2001 Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1 75 82
9. HochbergDMiddeldorpJMCatalinaMSullivanJLLuzuriagaK 2004 Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci U S A 101 239 244
10. JohannsenEMillerCLGrossmanSRKieffE 1996 EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-transformed B lymphocytes. J Virol 70 4179 4183
11. RobertsonESLinJKieffE 1996 The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J Virol 70 3068 3074
12. ZhaoBSampleCE 2000 Epstein-barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an spi-1/Spi-B binding site. J Virol 74 5151 5160
13. KnightJSLanKSubramanianCRobertsonES 2003 Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J Virol 77 4261 4272
14. RadkovSATouitouRBrehmARoweMWestM 1999 Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J Virol 73 5688 5697
15. SubramanianCHasanSRoweMHottigerMOrreR 2002 Epstein-Barr virus nuclear antigen 3C and prothymosin alpha interact with the p300 transcriptional coactivator at the CH1 and CH3/HAT domains and cooperate in regulation of transcription and histone acetylation. J Virol 76 4699 4708
16. SubramanianCCotterMA2ndRobertsonES 2001 Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: a molecular link to cancer metastasis. Nat Med 7 350 355
17. KnightJSSharmaNRobertsonES 2005 Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc Natl Acad Sci U S A 102 18562 18566
18. KnightJSSharmaNRobertsonES 2005 SCFSkp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol Cell Biol 25 1749 1763
19. KnightJSSharmaNKalmanDERobertsonES 2004 A cyclin-binding motif within the amino-terminal homology domain of EBNA3C binds cyclin A and modulates cyclin A-dependent kinase activity in Epstein-Barr virus-infected cells. J Virol 78 12857 12867
20. BajajBGMurakamiMCaiQVermaSCLanK 2008 Epstein-Barr virus nuclear antigen 3C interacts with and enhances the stability of the c-Myc oncoprotein. J Virol 82 4082 4090
21. SahaAMurakamiMKumarPBajajBSimsK 2009 Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J Virol 83 4652 4669
22. YiFSahaAMurakamiMKumarPKnightJS 2009 Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 388 236 247
23. CharrouxBPellizzoniLPerkinsonRAShevchenkoAMannM 1999 Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol 147 1181 1194
24. GrundhoffATKremmerETureciOGliedenAGindorfC 1999 Characterization of DP103, a novel DEAD box protein that binds to the Epstein-Barr virus nuclear proteins EBNA2 and EBNA3C. J Biol Chem 274 19136 19144
25. KrauerKGBuckMBelzerDKFlanaganJChojnowskiGM 2004 The Epstein-Barr virus nuclear antigen-6 protein co-localizes with EBNA-3 and survival of motor neurons protein. Virology 318 280 294
26. VossMDHilleABarthSSpurkAHennrichF 2001 Functional cooperation of Epstein-Barr virus nuclear antigen 2 and the survival motor neuron protein in transactivation of the viral LMP1 promoter. J Virol 75 11781 11790
27. CordinOBanroquesJTannerNKLinderP 2006 The DEAD-box protein family of RNA helicases. Gene 367 17 37
28. RocakSLinderP 2004 DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5 232 241
29. OuQMouilletJFYanXDornCCrawfordPA 2001 The DEAD box protein DP103 is a regulator of steroidogenic factor-1. Mol Endocrinol 15 69 79
30. YanXMouilletJFOuQSadovskyY 2003 A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol 23 414 423
31. GillianALSvarenJ 2004 The Ddx20/DP103 dead box protein represses transcriptional activation by Egr2/Krox-20. J Biol Chem 279 9056 9063
32. LeeKPisarskaMDKoJJKangYYoonS 2005 Transcriptional factor FOXL2 interacts with DP103 and induces apoptosis. Biochem Biophys Res Commun 336 876 881
33. KlappacherGWLunyakVVSykesDBSawka-VerhelleDSageJ 2002 An induced Ets repressor complex regulates growth arrest during terminal macrophage differentiation. Cell 109 169 180
34. SahaABamideleAMurakamiMRobertsonES 2010 EBNA3C attenuates the function of p53 through interaction with inhibitor of growth family proteins 4 and 5. J Virol 85 2079 2088
35. CauchiRJDaviesKELiuJL 2008 A motor function for the DEAD-box RNA helicase, Gemin3, in Drosophila. PLoS Genet 4 e1000265
36. MouilletJFYanXOuQJinLMugliaLJ 2008 DEAD-box protein-103 (DP103, Ddx20) is essential for early embryonic development and modulates ovarian morphology and function. Endocrinology 149 2168 2175
37. PellizzoniLCharrouxBRappsilberJMannMDreyfussG 2001 A functional interaction between the survival motor neuron complex and RNA polymerase II. J Cell Biol 152 75 85
38. FischerULiuQDreyfussG 1997 The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90 1023 1029
39. RossollWJablonkaSAndreassiCKroningAKKarleK 2003 Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163 801 812
40. YoungPJDayPMZhouJAndrophyEJMorrisGE 2002 A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy. J Biol Chem 277 2852 2859
41. HeinzelTLavinskyRMMullenTMSoderstromMLahertyCD 1997 A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 387 43 48
42. LeeMBLebedevaLASuzawaMWadekarSADesclozeauxM 2005 The DEAD-box protein DP103 (Ddx20 or Gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol Cell Biol 25 1879 1890
43. RenoufBHollvilleEPujalsATetaudCGaribalJ 2009 Activation of p53 by MDM2 antagonists has differential apoptotic effects on Epstein-Barr virus (EBV)-positive and EBV-negative Burkitt's lymphoma cells. Leukemia 23 1557 1563
44. PujalsARenoufBRobertAChelouahSHollvilleE 2011 Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis 2 e184
45. ForteELuftigMA 2009 MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected-cell survival. J Virol 83 2491 2499
46. O'NionsJTurnerACraigRAlldayMJ 2006 Epstein-Barr virus selectively deregulates DNA damage responses in normal B cells but has no detectable effect on regulation of the tumor suppressor p53. J Virol 80 12408 12413
47. CaiQLKnightJSVermaSCZaldPRobertsonES 2006 EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog 2 e116
48. AielloLGuilfoyleRHuebnerKWeinmannR 1979 Adenovirus 5 DNA sequences present and RNA sequences transcribed in transformed human embryo kidney cells (HEK-Ad-5 or 293). Virology 94 460 469
49. PontenJSakselaE 1967 Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer 2 434 447
50. ClementsGBKleinGPoveyS 1975 Production by EBV infection of an EBNA-positive subline from an EBNA-negative human lymphoma cell line without detectable EBV DNA. Int J Cancer 16 125 133
51. RobertsonESGrossmanSJohannsenEMillerCLinJ 1995 Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J Virol 69 3108 3116
52. KnightJSRobertsonES 2004 Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J Virol 78 1981 1991
53. LaemmliUK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680 685
54. DullTZuffereyRKellyMMandelRJNguyenM 1998 A third-generation lentivirus vector with a conditional packaging system. J Virol 72 8463 8471
55. WeiFZapraznaKWangJAtchisonML 2009 PU.1 can recruit BCL6 to DNA to repress gene expression in germinal center B cells. Mol Cell Biol 29 4612 4622
56. BaiYSrinivasanLPerkinsLAtchisonML 2005 Protein acetylation regulates both PU.1 transactivation and Ig kappa 3’ enhancer activity. J Immunol 175 5160 5169
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2011 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis