
Lineární IgA bulózní dermatóza. Popis případu
Linear IgA Bullous Dermatosis. Case Report
Linear IgA bullous dermatosis is a rare mucocutaneous autoimmune disorder, which belongs to a group of bullous diseases. The authors report a case of 85 year-old man with this disease confirmed by histopathology a direct immunofluorescence examination. Treatment with topical and oral corticosteroids induced remission of the disease. An overview of current knowledge of linear IgA dermatosis is provided.
Keywords:
linear IgA bullous dermatosis – bullous diseases
Autoři:
L. Lojkásková 1; L. Pock 2
Působiště autorů:
Kožní oddělení, Fakultní nemocnice Ostrava, přednosta prim. MUDr. Yvetta Vantuchová, Ph. D.
1; Bioptická laboratoř Plzeň, s. r. o., odborná vedoucí lékařka prof. MUDr. Alena Skálová, CSc.
2
Vyšlo v časopise:
Čes-slov Derm, 94, 2019, No. 5, p. 204-208
Kategorie:
Kazuistiky
Souhrn
Lineární IgA bulózní dermatóza je vzácná mukokutánní autoimunitní choroba, která patří do skupiny puchýřnatých onemocnění. Kazuistika popisuje případ 85letého pacienta, u kterého došlo k pozvolnému rozvoji projevů s následnou, rychle navozenou remisí lokálními a perorálními kortikosteroidy. Autoři uvádí přehled současných znalostí o tomto onemocnění.
Klíčová slova:
lineární IgA bulózní dermatóza – puchýřnatá onemocnění
ÚVOD
Lineární IgA bulózní dermatóza (LABD) je vzácné autoimunitní onemocnění s tvorbou subepidermálního puchýře. Incidence tohoto onemocnění se pohybuje mezi 0,25–2,3/1 000 000 obyvatel/rok [3, 4]. Obě pohlaví, bez ohledu na rasu, bývají postižena stejnou měrou. Rozlišujeme dvě klinické varianty onemocnění. Dětskou formu, která je známá též jako chronická bulózní dermatóza dětského věku, s věkovým průměrem výskytu 4,5 roku, a dospělou formu, pro kterou jsou typické dva věkové vrcholy onemocnění (16 let a 60 let). Klinický nález u tohoto onemocnění může být velmi různorodý, projevy se mohou vyskytovat na kůži i na sliznicích. Jsou popisovány rovněž asociace LABD navozené nádorovým onemocněním nebo vyvolané léky [5, 7, 14].
POPIS PŘÍPADU
Pacient, 85letý muž, byl přijat na kožní oddělení k diagnostické hospitalizaci a léčbě puchýřnatých projevů na těle a v dutině ústní. Pacient byl starobní důchodce, dříve pracoval jako chemik. Byl dispenzarizován na kardiologii (pro arteriální hypertenzi, stav po elektrické verzi flutteru síní, stav po infarktu myokardu, stav po koronárním bypassu s plastikou mitrální chlopně v roce 2002, stav po tranzitorní ischemické atace ve vertebrobazilárním povodí), interně (pro diabetes mellitus II. typu, hyperurikémii, chronickou venózní insuficienci s varixy dolních končetin, subklinickou hypothyreózu) a na urologii (pro hydrokélu levého varlete v roce 2012, léčené konzervativně, stav po transuretrální resekci prostaty (2004) pro benigní hyperplazii prostaty, papilokarcinom močového měchýře diagnostikovaném v březnu 2015, stav po transuretrální resekci papilokarcinomu v květnu 2015 bez nutnosti další terapie, bez známek recidivy po 24 měsících sledování).
Dlouhodobá léčba, neměnná po dobu 10 let, zahrnovala kyselinu acetylsalicylovou, trimetazidin, acebutolol, amiodaron, metformin, isosorbit mononitrát, levothyroxin a kombinovaný preparát perindoprilu s indapamidem.
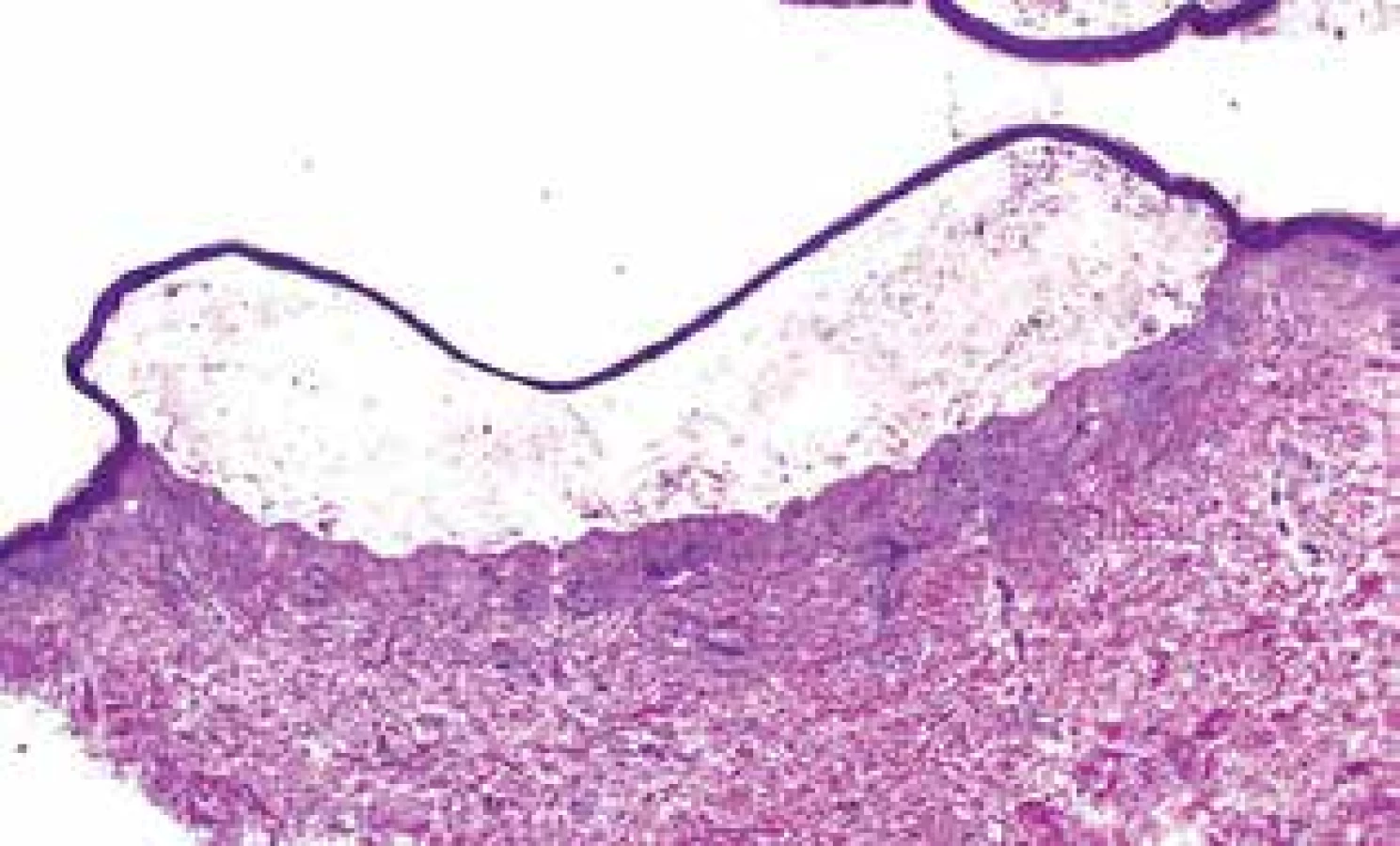
První projevy se objevily na podzim roku 2016 okrskovitým svěděním kůže se vznikem následného erytému a výsevu drobných čirých vezikul. Od února 2017 se vytvořily puchýřnaté projevy i na horním patře dutiny ústní. Výsevu nepředcházela infekce ani změna v chronické medikaci. Přímý i nepřímý Nikolského fenomén byl negativní. V den příjmu byly u pacienta přítomny vezikulózní projevy pouze v dutině ústní, v podpaží a tříslech, eroze po bulách, některé s hemoragickými krustami, byly přítomny v oblasti kštice, na bradě, krku a přední ploše trupu (obr. 1). Za hospitalizace byla odebrána základní laboratorní vyšetření, která byla bez výraznějšího patologického nálezu. Vzhledem k astenickému vzhledu pacienta a přítomnosti maligního onemocnění v anamnéze byl doplněn rentgen srdce a plic a ultrazvukové vyšetření břicha. Žádné z těchto vyšetření neprokázalo přítomnost solidního tumoru či jiné patologie. Močový měchýř byl na ultrazvuku popsán jako homogenní, ostře konturovaný. Histopatologické vyšetření prokázalo přítomnost subepidermální buly se zachovalou krytbou a zachovalým tvarem dermálních papil na spodině, s obsahem dosti četných polynukleárů a nečetných eozinofilů v horním koriu s perivaskulárně uloženými malými infiltráty lymfocytů s nečetnými eozinofily (obr. 2). V diferenciální diagnostice byl na prvním místě zvažován bulózní pemfigoid a lineární IgA dermatóza. Diagnóza IgA lineární dermatózy byla potvrzena metodou přímé imunofluorescence, kde byla přítomna silná lineární IgA pozitivita na bazální membráně epidermis. IgM, IgG, C3 byly negativní, s perivaskulárně pozitivními depozity fibrinogenu (obr. 3). Diagnostika metodou nepřímé imunofluorescence nebyla provedena. S přihlédnutím k celkovému stavu pacienta, jeho hmotnosti (64 kg), lokálnímu nálezu, který imitoval pemfigoid, chronické medikaci a komorbiditám byla zahájena léčba Prednisonem per os v celkové denní dávce 40 mg. Lokálně byla aplikována kortikosteroidní externa s obsahem antibiotik. Na základě této léčby došlo k postupné regresi projevů. Nové vezikuly se na těle již netvořily, stávající projevy se hojily. V dutině ústní vezikuly nejprve erodovaly a jejich hojení bez jizev bylo protrahované. Pacient byl propuštěn do ambulantní péče. Celková denní dávka Prednisonu byla postupně pomalu snižována až na 10 mg v průběhu 6 týdnů. Po tomto období byly projevy zcela zhojeny, k erupci nových nedošlo. Na další kontroly byl odeslán do spádové kožní ambulance, rovněž byl nadále dispenzarizován na urologii.


DISKUSE
Z epidemiologického hlediska je LABD vzácné autoimunitní bulózní onemocnění. Zatímco na kůži se projevy zhojují ad integrum, na sliznicích často dochází k jizvení. Toto onemocnění bylo poprvé popsáno v roce 1901 Bowenem, samostatnou jednotkou se však stalo až roku 1979. Klinicky rozlišujeme 2 formy LABD, dětskou a dospělou, které se liší věkovou distribucí a lokalizací projevů [6].
Dětská forma, neboli také chronická bulózní dermatóza dětského věku (CBCD), se objevuje v první dekádě života. Jedná se o nejčastější bulózní autoimunitní onemocnění dětského věku. Jednotlivé projevy bývají anulárně uspořádány a mají vzhled tzv. šňůry perel. Průměrná délka trvání CBDC je okolo 4 let. Jedná se o spontánně ustupující onemocnění, které u více než 50 % dětí odeznívá do 2 let od vzniku onemocnění.
U dospělé formy se na svědivé erytémové spodině objevují papuly a napjaté vezikuly či buly. Buly mohou samovolně praskat, a přeměnit se tak v eroze s krustami. Projevy LABD nemají predilekční lokalizaci, přesto se často objevují na stehnech, genitálu, v dolní části zad, ve kštici a na obličeji. Může být postižena sliznice spojivky, dutiny ústní a nosní, hltanu, jícnu či oblast konečníku. Pro postižení sliznic někteří preferují termín lineární IgA nemoc, ne dermatóza. Na rozdíl od dětské formy má tato forma protrahovanější průběh a procento remisí je nižší. Kožní změny se hojí bez jizvy, avšak na sliznicích k jizvení dojít může [2, 3, 6].
Diagnostika LABD se opírá především o dermatohistologické vyšetření. Mikroskopicky nacházíme subepidermální puchýř s dominantním neutrofilním infiltrátem v oblasti dermoepidermální junkce. Histopatologický nález je velmi podobný bulóznímu pemfigoidu, případně dermatitis herpetiformis. Zlatým standardem k potvrzení finální diagnózy je přímá imunofluorescence, kde jsou prokazována homogenní lineární IgA depozita podél bazální membrány epidermis. Vzácně zde může koexistovat pozitivita IgG, IgM a C3. Nepřímá imunofluorescence není prováděna rutinně. Touto metodou lze prokázat solubilní cirkulující IgA autoprotilátky v séru nemocných. Pozitivita nepřímé imunofluorescence bývá častěji u dětské formy [2, 3, 7, 8].
Klinická diferenciální diagnostika zahrnuje především bulózní pemfigoid a dermatitis herpetiformis, mimo jiné je pak také třeba zvážit bulózní impetigo a epidermolysis bullosa acquisita [1, 2, 4, 12]. Bulózní pemfigoid se klinicky projevuje bulami a erozemi, často bývají postiženy sliznice. Obvykle postihuje pacienty vyšších věkových skupin. Dermatohistopatologicky prokazujeme subepidermální puchýř se zvýšeným počtem eozinofilů. Přímá imunofluorescence zobrazuje lineární depozita IgG a C3 na bazální membráně. Dermatitis herpetiformis se projevuje seskupenými herpetiformně uspořádanými vezikulami v oblasti hlavy, sakra a extenzorů horních a dolních končetin. Na rozdíl od LABD je postižení sliznice dutiny ústní vzácné. Dermatohistopatologicky nacházíme neutrofilní mikroabscesy v oblasti dermálních papil, bez jejich lineárního řazení podél dermoepidermální junkce. Přímá imunofluorescence prokazuje zrnitou imunofluorescenci IgA s maximem ve vrcholcích papil koria. Jedná se o kožní manifestaci celiakie. U většiny pacientů kožní projevy dobře ustupují po zavedení bezlepkové diety. Bulózní impetigo vzniká častěji u dětí než u dospělých na podkladě infekce Staphylococcus aureus. Na obličeji, trupu a končetinách se tvoří erodující intraepidermální buly v oblasti stratum granulosum, které poté vytvářejí typické eroze s medovými krustami. Epidermolysis bullosa acqui-sita se od LABD odlišuje v přímé imunofluorescenci průkazem převážně depozit IgG na dermální straně puchýře. V některých případech může LABD imitovat prurigo nodularis, morbiliformní lékové erupce, erythema multiforme nebo toxickou epidermální nekrolýzu [4, 7, 12].
LABD byla v některých případech asociována s maligními tumory, zejména s lymfoproliferativními procesy (asi v 5%). Asociace mezi non-lymfoidními tumory a LABD nebyla dosud prokázána. Ojediněle byly v literatuře popsány případy viscerálních tumorů u pacientů s LABD, v případě karcinomu močového měchýře byly v minulosti popsány 4 případy [9, 14]. Obecně je však asociace mezi bulózním onemocněním a malignitou stále kontroverzním tématem. Vysoká incidence maligních onemocnění bývá uváděna u pacientů s pemfigem, epidermolysis bullosa acquisita a bulózním pemfigoidem. Nicméně i když byly tyto případy mnohokrát popsány, kontroverze přetrvávají. U LABD byla popsána asociace s autoimunitními chorobami (systémový lupus erythematodes, revmatoidní artritida, hypothyreóza, chronická hepatitida, IgA nefropatie, Crohnova choroba, ulcerózní kolitida), infekcemi (tuberkulóza, infekce horních dýchacích cest) a léky (nesteroidní antiflogistika, antibiotika – vankomycin, cefalosporiny, inhibitory angiotensin-konvertující-enzym). Patogeneze vzniku LABD v těchto případech není zcela objasněna, nicméně po léčbě stávajícího onemocnění či vysazení léku dochází k remisi do několika týdnů. Jedna z teorií vzniku LABD u pacientů s ulcerózní kolitidou je založena na předpokladu, že chronický zánět střev odhalí antigeny bazální membrány, jejich změnou konformace dojde ke vzniku neoantigenů, které stimulují slizniční imunitní systém k tvorbě autoprotilátek [2, 6, 9, 14].
Přesný patofyziologický proces vzniku onemocnění není dosud jasný. V patogenezi se uplatňuje buněčná i humorální imunitní odpověď, která vede k aktivaci komplementu, zánětlivé buněčné odpovědi a následné proteolýze. Na imunitní odpovědi se podílejí zejména lymfocyty (převládají pomocné CD4+ T lymfocyty), dále pak také neutrofily a eozinofily. Puchýře se tvoří sekundárně na základě poškození tkáně z důvodu lokalizované zánětlivé odpovědi, které se účastní neutrofily, degranulace žírných buněk a proteolytických enzymů uvolňovaných v procesu [7]. Základní patofyziologický mechanismus lze vysvětlit na solí děleném substrátu kůže (salt-split skin substrate, SSS). Inkubací tkáňového vzorku v 1 molárním roztoku NaCl dochází k arteficiálně navozenému štěpu v oblasti lamina lucida dermoepidermální junkční zóny. Na základě ultrastrukturálního rozmístění IgA autoprotilátek lze popsat 2 subtypy LABD [2, 7, 8, 10].
Prvním je subtyp lamina lucida bazální membrány epidermis, u které IgA autoprotilátky adherují k epidermální části štěpu SSS (neboli ke krytbě puchýře). IgA depozita jsou uložena v horní vrstvě lamina lucida a jsou ve spojení s membránou bazálních keratinocytů. Tato IgA depozita jsou uložena obdobně jako depozita IgG u bulózního pemfigoidu a společně s bulózním pemfigoidem sdílí také společné antigeny. IgA protilátky jsou zde namířeny proti adheznímu komplexu dermoepidermální junkce – 97 kDa antigenu a 120 kDa antigenu v lamina lucida. Tyto antigeny jsou fragmenty antigenu bulózního pemfigoidu 2 (BP180). Antigen 97 kDa je proteolytický fragment antigenu 120 kDa. Odštěpení ektodomény vede k indukci konformačních změn a vzniku nových epitopů, které se liší od epitopů rodičovského proteinu BPAg2 (180 kDa).
Druhý subtyp sublamina densa bazální membrány epidermis je oproti výše uvedenému subtypu méně častý a pacienti bývají rezistentní k léčbě sulfony. Autoprotilátky v séru zde reagují s dermální částí štěpu (neboli se spodinou puchýře). Depozita IgA jsou uložena podél kotvících fibril. U tohoto subtypu jsou predominantní autoantigeny 180 kDa, 285 kDa a kolagen VII [4, 13].
V léčbě LABD je první linií volby dapson, imunomodulační sulfon. U pacientů, kteří mají deficit glukosa-6-fosfát-dehydrogenázy, může dojít k rozvoji hemolytické anémie. U ostatních pacientů může dapson navodit methemoglobinémii, nebo se mohou objevovat známky počínající hemolýzy, která se projeví kompenzatorní retikulocytózou. Tito pacienti by pak mohli být spíše léčeni sulfapyridiny nebo sulfamethoxypyrazidiny, které jsou v ČR spolu s dapsonem dlouhodobě nedostupné a lze o ně zažádat formou mimořádného dovozu. Jedná se o sulfonamidy, které s dapsonem sdílejí podobnou strukturu, avšak riziko nežádoucích účinků je nižší. Terapii zahajujeme podáním sulfonů v dávce 25–50 mg/den s postupným navýšením dávky na 100–150 mg/den, u dětí nepřekračujeme dávku 0,5 mg/kg/den. Před zahájením léčby sulfony je vhodné stanovit hladinu glukosa-6-fosfát-dehydrogenázy v séru pacienta. Zlepšení klinického nálezu lze očekávat do 3 dnů. Sulfapyridin lze podat v dávce od 250 mg/den do 3 g/den. V případě selhání první linie jsou celkově podávány kortikosteroidy v dávce až 1 mg/kg/den, a to samostatně či v kombinaci s jinými imunosupresivy. Náš pacient byl léčen kortikosteroidy, neboť klinický nález imponoval jako pemfigoid a výsledek histologického vyšetření nebyl v době zahájení léčby k dispozici. Na základě klinicko-histo-patologické korelace byla stanovena diagnóza LABD a kortikosteroidy, které byly účinné, byly pacientovi ponechány. V literatuře jsou popisováni i pacienti, kteří byli léčení kolchicinem (především děti), azathioprinem, rituximabem či kombinací s tetracykliny a niacinamidem. Samozřejmě, je-li onemocnění indukováno léky, stačí prosté vysazení tohoto léku [1, 2, 10, 11, 12].
ZÁVĚR
Námi demonstrovaný případ ukazuje pacienta s LABD nejasné etiologie. Za úvahu by u tohoto pacienta stála i poléková etiologie, nicméně pacient v době vzniku onemocnění zavedenou medikaci užíval dlouhodobě. V případě lékové etiologie vznikají příznaky do 1 měsíce od počátku užívání léku, v některých případech v delším časovém odstupu od zahájení léčby. Jiné interní onemocnění či malignitu ve vztahu k LABD jsme u pacienta neprokázali.
Ke stanovení diagnózy je potřebné nejen dermatohistopatologické vyšetření, ale vzhledem k příbuznosti jiných bulózních dermatóz i přímá imunofluorescence, která nám prokáže lineární IgA depozita podél bazální membrány.
U pacientů může po nějaké době (většinou do 2 let) dojít ke spontánní remisi onemocnění. U jiných pacien-tů může onemocnění přetrvávat i desetiletí, stejně tak může docházet k recidivám.
Prohlašuji, že jsem v souvislosti s tématem této práce v posledních 12 měsících nespolupracovala s žádnou farmaceutickou firmou.
Do redakce došlo dne 3. 9. 2019.
Adresa pro korespondenci
MUDr. Lenka Lojkásková
Kožní oddělení
Fakultní nemocnice Ostrava
17. listopadu 1790/5
708 52 Ostrava
e-mail: lenka.lojkaskova@fno.cz
Zdroje
1. BRAUN-FALCO, O., WOLFF, H., PLEWIG, G. Braun-Falco’s Dermatologie, Venerologie und Allergologie. Springer, Verlag Berlin Heidelberg, 2018. ISBN 978-3-662-49543-8, p. 861
2. CURTIS, J. A., JOHN J. Z. Linear IgA bullous dermatosis. In Naveed, S. Autoimmune Bullous Diseases. Cham: Springer International Publishing, 2016, p. 117–132. DOI: 10.1007/978-3-319-26728-9_7. ISBN 978-3-319-26726-5. Dostupné na www: http://link.springer.com/10.1007/978-3-319-26728-9_7.
3. FERNANDES, K. de A. P. et al. Linear IgA and IgG bullous dermatosis. An. Bras. Dermatol., 2016, 91(5, suppl.1), pp.32–34. ISSN 0365-0596. Dostupné na www: http://dx.doi.org/10.1590/abd1806-4841.20164630.
4. FORTUNA, G., MARINKOVICH, P. M. Linear immunoglobulin A bullous dermatosis. Clinics in Dermatology, 2012, 30(1), p. 38–50. ISSN 0738-081X. Dostupné na www: https://doi.org/10.1016/j.clindermatol.2011.03.008.
5. GENOVESE, G., VENEGONI, L., FANONI, D., MURATORI, S., BERTI, E., MARZANO, A.V. Linear IgA bullous dermatosis in adults and children: a clinical and immunopathological study of 38 patients. Orphanet J Rare Dis., 2019, 14(1), p. 115. DOI:10.1186/s13023-019-1089-2.
6. HAEBERLE, M. T., CALLEN, J. P. Linear IgA Dermatosis. 2017. Dostupné na www: http://emedicine.medscape.com/article/1063590-overview.
7. CHAUDHARI, S., MOBINI, N. Linear IgA Bullous Dermatosis: A Rare Clinicopathologic Entity with an Unusual Presentation. The Journal of Clinical and Aesthetic Dermatology, 8(10), p. 43–46.
8. OTTEN, J. V., HASHIMOTO, T., HERTL, M., PAYNE A. S., SITARU C. Molecular Diagnosis in Autoimmune Skin Blistering Conditions. Current Molecular Medicine, 2014, 14(1), p. 69–95. DOI: 10.2174/15665240113136660079. Dostupné na www: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1566-5240&volume=14&issue=1&spage=69.
9. RODENAS, J. M., HERRANZ, M. T., TERCEDOR, J., CONCHA, A. Linear IgA disease in a patient with bladder carcinoma. British Journal of Dermatology, 1997, 136(2), p.257–259. DOI: 10.1046/j.1365-2133.1997.d01-1182.x.
10. ROOK, A. J., BARKER, J., BLEIKER, T., CHALMERS, R., CREAMER, D., GRIFFITHS, C. E. Immunobullous Diseases. In Rooks textbook of dermatology (9th ed., Vol. 4). Chichester, West Sussex (UK): Wiley Blackwell, 2016. p. 50.33–50.38.
11. SALAVEC, M., BOŠTÍKOVÁ, N. Sulfony v dermatologii. Čes-slov. Derm., 2017, 92(4), p. 155–175.
12. ŠTORK, J. Dermatovenerologie. Česká republika: Galén, 2013, p. 210.
13. UTSUNOMIYA, N., CHINO, T., OYAMA, N., UTSUNOMIYA, A., YAMAGUCHI, Y., TAKASHIMA, W. et al. Sublamina densa-type linear IgA bullous dermatosis with IgA autoantibodies specific for type VII collagen: a case report and clinicopathological review of 32 cases. Dermatology Online Journal, 2017, 23(11).
14. VAN DER WAAL, R. I., VAN DE SCHEUR, M. R., PAS, H. H., JONKMAN, M., VAN GROENINGEN, C. J., NIEBOER, C., STARINK, T. M. Linear IgA bullous dermatosis in a patient with renal cell carcinoma. British Journal of Dermatology, 2001, 144(4), p.870–873. DOI: 10.1046/j.1365-2133.2001.04148.x.
Štítky
Dermatológia Detská dermatológiaČlánok vyšiel v časopise
Česko-slovenská dermatologie

2019 Číslo 5
- Na český trh přichází biosimilar adalimumabu s prokázanou terapeutickou ekvivalencí
- Nehoňte nemocné s mMCC od čerta k ďáblu!
- První a jediná schválená imunoterapie vzácného agresivního karcinomu kůže
- Screening malignit u pacientů s dermatomyozitidou
- Jakým způsobem hydroresponzivní krytí napomáhá hojení rány?
Najčítanejšie v tomto čísle
- Kožní nádory
- Liekové hypersenzitívne reakcie: diagnostika a liečba (2. časť)
- Lineární IgA bulózní dermatóza. Popis případu
- Průsvitná papula na prstu nohy. Stručný přehled