
Alagilleův syndrom
Alagille syndrome
Alagille syndrome – AGS (OMIM 118450) is a highly variable multisystem autosomal dominant disorder with incomplete penetrance. The disease is caused by mutations in JAG1 or rarely in NOTCH2. Both genes encode extracellular signaling proteins involved in embryogenesis. Presence of pathogenic mutations leads to multiple malformations. AGS has extremely pleitropic clinical manifestations, even within the same family members carrying the same mutations. Consequentely, there is no correlation between genotype and phenotype.
Cholestatic liver disease due to bile duct paucity with high levels of cholesterol, formation of xanthomas and a rigorous pruritus, cardiovascular anomalies (most commonly peripheral pulmonary artery stenosis), “butterfly-like” vertebrae, ophtalmologic anomalies (posterior embrytoxon) and a typical craniofacial dysmorphia including triangular face, prominent forehead, saddle nose and narrow chin represent the key clinical features of AGS. Renal, vascular and other malformations are also frequent. Liver transplantation in childhood is necessary in cases with severe liver disease.
Key words:
Alagille syndrome, bile duct paucity, JAG1, neonatal cholestasis
Autoři:
T. Dědič 1; M. Jirsa 2; R. Kotalová 1
Působiště autorů:
Pediatrická klinika UK 2. LF a FN Motol, Praha, přednosta prof. MUDr. J. Lebl, CSc.
1; Laboratoř experimentální hepatologie IKEM, Prahavedoucí doc. MUDr. Mgr. M. Jirsa, CSc.
2
Vyšlo v časopise:
Čes-slov Pediat 2014; 69 (4): 241-249.
Kategorie:
Přehledový článek
Souhrn
Alagilleův syndrom – AGS (OMIM 118450) je vysoce variabilní multisystémové autozomálně dominantně dědičné onemocnění s neúplnou penetrancí. Onemocnění vyvolávají mutace v genech JAG1 nebo vzácně NOTCH2. Oba geny kódují mezibuněčné signální proteiny uplatňující se v embryogenezi, přítomnost patogenních mutací podmiňuje mnohočetné malformace. AGS má extrémně variabilní projevy, a to i v rámci jedné rodiny, neboť neexistuje korelace mezi genotypem a fenotypem.
AGS charakterizuje cholestatické jaterní onemocnění – duktopenie intrahepatálních žlučových cest provázená cholestázou s vysokými hodnotami cholesterolu, tvorbou xantomů a úporným pruritem, kardiovaskulární anomálie (nejčastěji stenózy periferních větví plicnice), motýlovité obratle, kongenitální oční anomálie (embryotoxon posterior) a typická kraniofaciální dysmorfie zahrnující trojúhelníkovitý obličej, prominující čelo, sedlovitý nos a úzkou bradu. Časté jsou renální, cévní a jiné malformace. U některých jedinců je nezbytná transplantace jater již v dětství.
Klíčová slova:
Alagilleův syndrom, duktopenie, JAG1, neonatální cholestáza
Úvod
Alagilleův syndrom (OMIM 118450) je multisystémové autozomálně dominantně dědičné onemocnění dané poruchou mezibuněčné signální dráhy NOTCH [1]. První klinický případ byl popsán v roce 1969 D. Alagillem [2].
Prevalence v populaci (včetně neúplně vyjádřených a atypických forem) je odhadována na 1 : 30 000. Incidence na podkladě neonatální cholestázy se pohybuje kolem 1 : 70 000, ale je téměř jisté, že toto číslo je podhodnocené pro variabilitu klinických příznaků a neúplnou penetranci onemocnění [3].
Molekulární podstata
AGS je autozomálně dominantní geneticky heterogenní onemocnění s vysoce variabilní penetrancí i expresivitou, které je způsobené mutací v genu JAG1 (AGS 1. typu, OMIM #118450) [4, 5] u 94 % všech probandů) nebo vzácně v genu NOTCH2 (AGS 2. typu, OMIM #610205) [6] u 1 % probandů). Gen JAG1 sestává z 26 exonů, nachází se na krátkém raménku 20. chromozomu (20p12) a kóduje povrchový protein Jagged1, který se váže jako ligand na transmembránové receptory rodiny Notch (Notch1, 2, 3 a 4). Po navázání ligandu receptorem dochází ke spuštění signální dráhy Notch, jež hraje hlavní roli v diferenciaci kmenových buněk a embryogenezi. Protein Jagged1 se skládá ze 3 domén: velké extracelulární domény obsahující signální peptid (SP), DSL oblast odpovědnou za interakci s receptorem Notch, 16 EGF-like oblast, dále na cystein bohatou oblast. Poté následuje transmembránová doména zakončená krátkou intracelulární doménou.
Navázání ligandu Jagged1 vyvolá konformační změnu receptoru NOTCH2, která umožní odštěpení intercelulární domény NICD receptoru NOTCH2 metaloproteinázou. NICD přechází z membrány do cytoplazmy, kde interaguje s transkripčním faktorem CBF1. Vazbou NICD a CBF1 se aktivují další transkripční regulátory. Výsledkem těchto interakcí je tvorba iniciačního komplexu pro transkripci cílových genů. Cílovými geny signální dráhy NOTCH2 jsou geny z rodiny Hes a Hey. Tyto geny jsou zapojeny v embryogenezi kardiovaskulárního systému, jater a jiných systémů [4, 5].
Podle posledních prací 94 % probandů s AGS má mutaci v genu JAG1 [7]. Mutací bylo již popsáno přes 430 [11]. Většinou se jedná o bodové mutace (88 %), které způsobují předčasné ukončení translace (nonsense mutace) nebo posun čtecího rámce s následným předčasným ukončením translace (frameshift mutace), nebo missense mutace (11 %). Přibližně 7 % pacientů může nést mikrodeleci krátkého raménka chromozomu 20 [8]. Asi 50–70 % mutací v JAG1 vzniká de novo, pouze ve 30–50 % je mutace děděna po matce či otci. Pravděpodobným mechanismem působení mutací v JAG1 je haploinsuficience – snížení množství funkčního proteinu Jagged1 [9].
Gen NOTCH2 je tvořený 34 exony a nachází se na chromozomu 1 (1p13-p11). Je známo pouze 10 pacientů s mutací v genu NOTCH2 [10].
Klinické příznaky
AGS má morfologické změny především v játrech, kardiovaskulárním systému, očích a skeletu, charakteristická je též kraniofa-ciální dysmorfie. Časté jsou i anomálie ledvin, pankreatu a malý vzrůst. Četnost klinických příznaků v literárních souborech pacientů s AGS je uvedena v tabulce 1 [11].
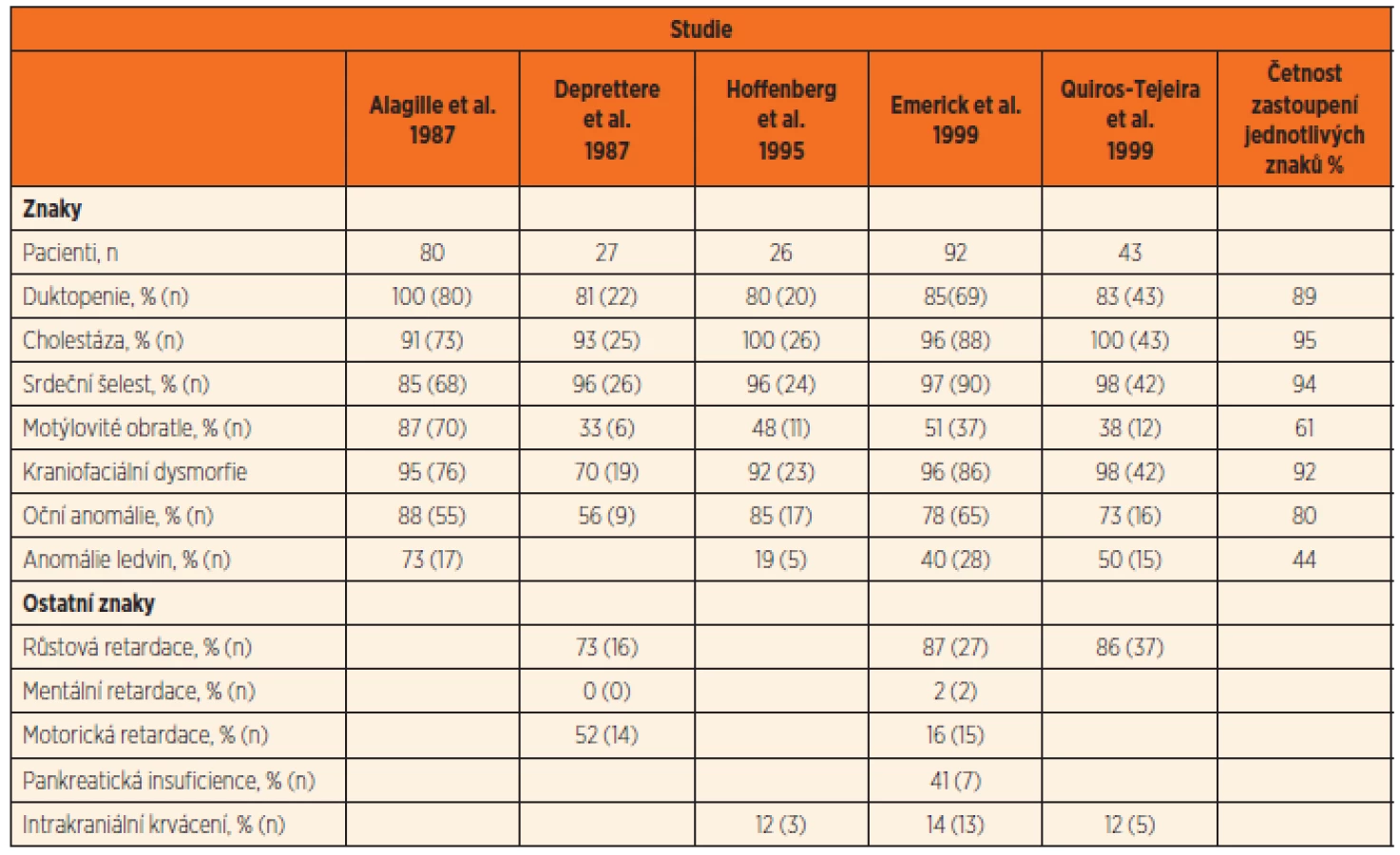
Morbidita a mortalita AGS je ovlivněna především závažností jaterního a kardiovaskulárního postižení.
Jaterní onemocnění
Příčinou jaterního onemocnění u AGS je duktopenie intrahepatálních žlučovodů označovaná též jako arteriohepatická dysplazie nebo intrahepatická dysgeneze. Klíčovou roli ve vývoji žlučových cest zde hraje gen JAG1.
Duktopenie je považována za základní znak syndromu. Obvyklý poměr počtu žlučových kanálků na portální pole je ve zdravých játrech 0,9–1,8. U dětí s AGS je tento poměr pouze 0,5–0,75 [2]. Ke zjištění poměru je potřebná biopsie zahrnující alespoň 10 portálních polí. Základním předpokladem pro stanovení histologické diagnózy je získání dostatečného množství jaterní tkáně. To je v časném kojeneckém věku obtížné, neboť odebrání reprezentativního vzorku nezaručuje ani jehlová biopsie, ani případná klínovitá subkapsulární resekce provedená při chirurgickém výkonu.
Velké studie prokazují duktopenii intrahepatálních žlučových cest u 75–100 % případů AGS [12–16]. V bioptickém materiálu bývá duktopenie přítomna u 95 % kojenců s AGS starších 6 měsíců, zatímco ve věku do 6 měsíců je prokazatelná pouze v 60 % reprezentativních biopsií [14].
Vedle AGS může být duktopenie v portálních polích zastižena i u neonatální hepatitidy, biliární atrézie a u některých metabolických a imunitních poruch. Její hodnocení z jedné biopsie u kojence je obtížné a nezřídka zavádějící.
Duktopenie intrahepatálních žlučových cest u AGS má variabilní klinický obraz. Asi pětina pacientů má jen minimální nebo žádné známky jaterního onemocnění. Většina pacientů s jaterním postižením se manifestuje v prvním roce života [12] jako ikterus s konjugovanou hyperbilirubinemií, hepatomegalií a hypocholickými až acholickými stolicemi s následným rozvojem malnutrice, osteoporózy a pruritu. Později se objevují xantomy – nejčastěji na extenzorových částech prstů, v oblasti zátylku, v podkolení, na hýždích a v tříslech. V laboratorním nálezu je vyjádřena cholestáza – elevace žlučových kyselin, konjugovaného bilirubinu, alkalické fosfatázy, gama-glutamyl transferázy (GGT) a vysoké hladiny cholesterolu a triacylglycerolů.
U 10–50 % pacientů, u kterých se objeví známky jaterního postižení již v kojeneckém věku, dochází k fibrotickým změnám až cirhotické přestavbě a portální hypertenzi nebo k rozvoji chronického jaterního selhání [13]. Etiopatogeneze chronického jaterního selhání se syntetickou dysfunkcí u AGS není plně objasněna. Závažnost stupně cholestázy v kojeneckém věku však není dobrým predikčním ukazatelem rozvoje chronického jaterního selhání.
Hypoplazie může postihnout i velké mimojaterní žlučovody, takže fenotyp AGS může napodobit biliární atrézii (BA). Nehledě na různost příčin se obě jednotky projeví v novorozeneckém období stejným klinickým a laboratorním nálezem. Rozlišení může dále komplikovat i neúplné vyjádření dalších znaků AGS [17]. Při cholescintigrafii, ERCP a dokonce ani při peroperační cholangiografii nemusejí být extrahepatální žlučové cesty dobře zobrazeny [6]. Jaterní biopsie je proto primární diagnostickou metodou. Včasné rozlišení obou jednotek má zásadní význam pro léčbu, neboť Kasaiova operace klinický stav pa-cientů s AGS nezlepšuje. U většiny operovaných naopak dochází k jeho zhoršení, takže u 60 % případů je nutno přikročit k transplantaci jater [17].
Je známo, že u pacientů s AGS se může vyskytnout hepatocelulární karcinom [18].
Srdeční onemocnění
Kardiovaskulární onemocnění je popisováno u 85–98 % pacientů s AGS [12–16, 19]. Nejčastěji se anomálie týkají pulmonální arterie a jsou to především mnohočetné stenózy jejích periferních větví (69–76 % pacientů). Ty jsou přítomny samostatně nebo v kombinaci [20]. Fallotova tetralogie patří mezi nejčastější komplexní srdeční vady vyskytující se u 7–12 % případů AGS [2, 13, 19]. Závažné formy Fallotovy tetralogie kombinované s atrézií pulmonální arterie jsou mnohem častější u pacientů s AGS (40 %) než v běžné populaci [13, 19]. Podle práce Emericka et al. [13] z jejich souboru 92 pacientů s AGS přibližně 11 % muselo podstoupit kardiochirurgickou intervenci. Mortalita u pacientů s Fallotovou tetralogií činila 33 % a u pacientů s Fallotovou tetralogii kombinovanou s atrézií plicnice až 75 %. Pacienti s AGS a těžkým srdečním postižením mají pouze 40% přežití do 6 let věku oproti 95% přežití u pacientů s AGS bez srdečního postižení [13].
Dále byly u pacientů s AGS popsány defekty septa komor a síní, aortální stenózy, koartace aorty, truncus arteriosus, hypoplazie levého srdce a další anomálie.
Srdeční onemocnění může být jediný příznak u pacientů AGS [13]. Mutace v genu JAG1 byly nalezeny u rodin v několika generacích, které měly pouze stenózy periferních větví plicnice bez jaterního onemocnění [21].
Anomálie skeletu
Malformace obratlů byly popsány jako jedno z hlavních diagnostických kritérií [2]. Jedná se o motýlovitý tvar obratlů vyskytující se u 33–87 % probandů [12–16]. U běžné populace se tato vývojová anomálie nevyskytuje, ale může být nalezena u jiných onemocnění, např. tzv. VATER asociace (asociace vertebrálních defektů, atrézie anu, tracheoezofageální píštěle, ezofageální atrézie a radiální a renální dysplazie), Crouzonova syndromu, Jarcho-Levin spondylokostální dysostózy, Kabuki syndromu a dalších. Motýlovité obratle jsou většinou asymptomatické. U AGS se mohou vyskytnout i jiné anomálie skeletu jako hemivertebra, absence 12. žebra, spina bifida occulta, krátké prsty se širokým palcem.
AGS bývá provázen závažnými osteoporotickými změnami, časté jsou patologické fraktury zvláště femuru [22]. K osteoporóze přispívá u AGS řada faktorů – chronická jaterní léze s následným sekundárním malabsorpčním syndromem provázená malnutricí s deficiencí vitaminů D a K, renální léze a pankreatická insuficience.
Oční anomálie
U pacientů s AGS byly popsány různé vývojové anomálie oka tykající se rohovky, duhovky, sítnice a optického nervu. Ty většinou neovlivní zrak, ale jsou důležitým diagnostickým znakem. Embryotoxon posterior je považován za jeden z typických diagnostických znaků AGS, který se vyskytuje u 56–95 %pacientů s AGS [23]. Jedná se o vývojovou anomálii charakterizovanou bílou linií v periferii rohovky lehce vystupující nad niveau – přední a prominující Schwalbeho linii. Tato anomálie může být přítomna u 15–22 % fyzio-logických očí [24]. U pacientů, kde se zadní embryotoxon vyskytuje samostatně, není zvýšené riziko glaukomu.
Druhou nejčastější oční anomálií je malformace optického disku přítomná alespoň na jednom oku. Dále se může vyskytnout kongenitální makulární dystrofie, extrofie či entropie čočky, microcornea, hypoplazie čočky a další [25].
Kraniofaciální dysmorfie
Typická kraniofaciální dysmorfie je považována za jedno z nejvýznamnějších diagnostických kritérií. Pacienti s AGS mají trojúhelnikovitý obličej s hypertelorismem, vpadlé oči, sedlovitý nos a prominující úzkou bradu [2]. Facies nebývá dobře rozpoznatelná u kojenců, ale s přibývajícím věkem nabývá charakteristických rysů. Je také zřejmá u symptomatických sourozenců a rodičů. Souvislost facies s expresí genu JAG1 byla prokázána i experimentálně na myším modelu, kde byla zjištěna exprese JAG1 při vývoji obličejových kostí myši [26]. Prevalence kraniofaciální dysmorfie u jedinců s AGS je 70–95 % [12–16].


Anomálie cévního systému a jeho komplikace
Intrakraniální krvácení je zaznamenáváno přibližně u 15 % pacientů s AGS [13, 16]. Ve většině případů se jedná o spontánní krvácení, které se v některých případech objevilo po mírném kraniotraumatu bez koagulační poruchy, bez trombocytopenie a jaterního selhání. V 30–50 % je krvácení fatální. Podkladem spontánního krvácení jsou abnormality cévního systému CNS (aneuryzma bazilární arterie nebo a. carotis media). Emerick [6] provedl prospektivně MR angiografii u 26 pacientů s AGS bez neurologické symptomatologie. U 10 (38 %) z nich nalezl abnormality cévního systému CNS. Na podkladě těchto zjištění se doporučuje u všech pacientů s AGS provést MR angiografii v rámci screeningu cévních změn CNS. U AGS byly také popsány anomálie a. carotis interna [27] a opakovaně byla zaznamenána moyamoya nemoc (progresivní intrakraniální arteriální okluzivní nemoc) [28]. Je pravděpodobné, že signální dráha NOTCH hraje jednu z klíčových rolí ve vývoji cévního systému.
Anomálie ledvin
Studie prokázaly, že 19–73 % pacientů s AGS má onemocnění ledvin [12–16] (tab. 1). Jedná se o různé strukturální či funkční anomálie – solitární ledvinu, ektopickou ledvinu dysplastické ledviny a anomálie ureteru či pánvičky. U některých probandů s AGS byla zaznamenána i polycystická postižení ledvin [29] s nebo bez renální insuficience, dále stenóza renální arterie jako příčina systémové hypertenze [30] a juvenilní nefronoftíza. Je též popisována renální tubulární acidóza a lipidóza glomerulů [31]. Renální selhání bylo indikací k transplantaci ledviny [15, 32]. Nedávná práce ukázala, že pacienti s mutací v genu NOTCH2 mají vyšší prevalenci postižení ledvin oproti pacientům s mutací v genu JAG1 [32].
Růstová retardace
Kolem 50–87 % pacientů s AGS má závažnou růstovou retardaci, která je zejména zřetelná v prvních 4 letech života [12–16]. Jedním z hlavních důvodů malnutrice je malabsorpce při cholestáze. Olsen [33] zjistil u pacientů s AGS signifikantní deficit ve velikosti a vlastnostech kostí a zvažuje možnost jejich vzniku na podkladě malabsorpce tuků. Je také možné, že tento deficit je důsledkem vývojových anomálií při AGS. Pacienti s AGS jsou rezistentní na terapii růstovým hormonem [34]. Pokud je dítě s AGS transplantováno, je pozorován určitý catch up po transplantaci, ale lineární růstový deficit přetrvává [35].
Pankreatické změny
Chong [36] popsal u některých pacientů s AGS s průjmy nízké hladiny lipázy v duodenální šťávě. Také byl u syndromu zaznamenán inzulin dependentní diabetes mellitus. Post mortem byly prokázány fibrotické změny pankreatu a abnormality pankreatických vývodů. Není jednoznačné, zda se jedná o primární změny nebo důsledek cholestázy s následnými metabolickými a toxickými změnami.
Jiné anomálie
U AGS byly popsány i jiné anomálie – stenózy trachey a bronchů, atrézie jejuna, ilea, malrotace střevní a mikrokolon, makrocefalie, striktury uretry, hypotyreóza a další.
Morbidita a mortalita
Mortalita je u AGS velmi variabilní. Nejčastější příčinou úmrtí jsou srdeční vady, dále jaterní léze a krvácení do CNS při cévních anomáliích. Závažné srdeční vady jsou nejčastější příčinou úmrtí u malých dětí. Úmrtí na jaterní selhání a krvácení do CNS přicházejí spíše později.
Transplantace jater je nutná asi u 20–50 % pacientů s AGS. Indikací k transplantaci jsou zejména progredující chronické jaterní selhání, komplikace portální hypertenze, ale i důsledky cholestázy – patologické fraktury, pruritus a růstová retardace. Jednoroční přežití po transplantaci je ve srovnání s jinými diagnózami nižší (87 %, u BA 96 %). Největší počet úmrtí je do 30 dnů po transplantaci [35]. V příčinách časných úmrtí dominují cévní komplikace. Situaci dále ovlivní přítomnost srdeční vady a stav ledvin.
Diagnostika AGS
Původní Alagillem navržená diagnostika syndromu stála na průkazu duktopenie intrahepatálních žlučových cest, která je provázena 3 z 5 možných dalších znaků, resp. kritérií syndromu: cholestáza, typická kraniofaciální dysmorfie, anomálie obratlů, oka a srdeční vada. S následným pozná-ním postižení dalších systémů bývají do základních kritérií přiřazeny i renální anomálie.
Molekulární diagnostika AGS stojí na průkazu mutací v genu JAG1 nebo NOTCH2. I přes současné možnosti exaktní genetické diagnostiky stále existuje určité procento jedinců, kteří dobře splňují základní kritéria AGS, ale mutace v uvedených genech nejsou nalezeny. Tyto jedince však lze považovat za nositele AGS. Naopak je známo, že u řady asymptomatických příbuzných probanda s AGS jsou prokazovány patogenní mutace v JAG1 nebo NOTCH2. Tyto skutečnosti vedly k definici tzv. revidovaných kritérií AGS [11] – viz tabulka 2.

Terapie
Léčba pacientů s AGS je i přes lepší poznání patofyziologie onemocnění stále především symptomatická. Jednou z hlavních klinických obtíží je cholestáza. Tok žluči je podporován choleretickým efektem léčebně podávané ursodeoxycholové kyseliny. Ta může do určité míry ovlivnit i pruritus. Obvykle je však k jeho řešení potřebné podání cholestyraminu nebo rifampicinu. Běžně, avšak bez většího efektu, jsou podávána antihistaminika. V pediatrii je jen výjimečně indikováno užívání naltrexonu [37]. U pacientů s přetrvávajícím úporným pruritem lze zvážit zajištění chirurgické drenáže žlučových cest cestou biliární diverze [38].
Vzhledem k malabsorpci při závažné cholestáze a následné růstové retardaci a malnutrici, osteoporóze a dalším situacím musí být u pacientů s AGS zajištěna optimální výživa. Kromě zvýšeného celkového příjmu kalorií je tuky nutno podávat ve formě triacylglycerolů se středními řetězci. Dále je nutná monitorace hladin vitaminů rozpustných v tucích a jejich substituce, stejně tak vápníku, železa, hořčíku a dalších prvků.
Konečné stadium jaterního onemocnění je řešeno transplantací jater. K transplantaci může přivést i neúnosný pruritus nereagující na medikamentózní terapii nebo opakující se patologické fraktury i v situaci bez pokročilého jaterního selhání. Současně je nutná léčba mimojaterních manifestací syndromu.
Podpořeno grantem GAUK č. 630512.
Došlo: 18. 3. 2014
Přijato: 20. 5. 2014
MUDr. Tomáš Dědič
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: Tomas.Dedic@fnmotol.cz
Zdroje
1. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling cell fate control and signal integration in development. Science 1999; 284 : 770–776.
2. Alagille D, Odièvre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr 1975; 86 : 63–71.
3. Kamath BM, Bason L, Piccoli DA, et al. Consequences of JAG1 mutations. J Med Genet 2003; 40 : 891–895.
4. Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16 : 243–251.
5. Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 1997; 16 : 235–242.
6. McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the Notch signaling pathway. Am J Hum Genet 2006; 79 : 169–173.
7. Warthen DM, Moore EC, Kamath BM, et al. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat 2006; 27 : 436–443.
8. Boyer J, Crosnier C, Driancourt C, et al. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Human Genet 2005; 116 : 445–453.
9. Morrissette JD, Colliton RP, Spinner NB. Defective intracelular transport and processing of JAG1 missence mutations in Alagille syndrome. Hum Mol Genet 2001; 10 : 405–413.
10. Kamath BM, Bauer RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet A 2012; 49 : 138–144.
11. Kamath BM, Spinner NB, Piccoli DA. Alagille syndrome. In: Suchy P (ed).: Liver Disease in Children. 3rd ed. New York: Cambridge University Press, 2007 : 326–345.
12. Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr 1987; 110 : 195–200.
13. Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999; 29 : 822–829.
14. Deprettere A, Portmann B, Mowat AP. Syndromic paucity of the intrahepatic bile ducts: diagnostic difficulty; severe morbidity throughout early childhood. J Pediatr Gastroenterol Nutr 1987; 6 : 865–871.
15. Quiros-Tejeira RE, Ament ME, Heyman MB, et al. Variable morbidity in Alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr 1999; 84 : 56–60.
16. Hoffenberg EJ, Narkewizc MR, Sondheimer JM, et al. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr 1995; 127 : 220–224.
17. Kaye AJ, Rand EB, Munoz PS, et al. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr 2010; 51 : 319–321.
18. Kaufmann SS, Wood RP, Shaw BW Jr, et al. Hepatocarcinoma in a child with the Alagille syndrome. Am J Dis Child 1987; 141 : 698–700.
19. McElhinney DB, Krantz ID, Bason L, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation 2002; 106 : 2567–2574.
20. Watson GH, Miller V. Arterio-hepatic dysplasia. Familial pulmonary arterial stenosis with neonatal liver diseases. Arch Dis Child 1973; 48 : 459–466.
21. Krantz ID, Smith R, Colliton RP, et al. Jagged1 mutations in patients ascertained with isolated congenital heart defects. Am J Med Genet 1999; 84 : 56–60.
22. Bales CB, Kamath BM, Munoz PS, et al. Pathologic lower extremity fractures in children with Alagille syndrome. J Pediatr Gastroenterol Nutr 2010; 51 : 66–70.
23. Hingorani M, Nischal KK, Davies A, et al. Ocular abnormalities in Alagille syndrome. Ophtalmology 1999; 106 : 330–337.
24. Rennie CA, Chowdhury S, Khan J, et al. The prevalence and associated features of posterior embryotoxon in the general ophthalmic cloníc. Eye 2005; 19 : 396–399.
25. Brodsky MC, Cunniff C. Ocular anomalies in the Alagille syndrome (arteriohepatic dysplasia). Ophthalmology 1993; 100 : 1767–1774.
26. Kamath BM, Loomes KM, Oakey RJ, et al. Facial features in Alagille syndrome: Specific or Alagille syndrome. Am J Med Genet 2002; 112 : 163–170.
27. Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: significant cause of morbidity and mortality. Circulation 2004; 109 : 1354–1358.
28. Woolfenden AR, Albers GW, Steinberg GK, et al. Moyamoya syndrome in children with Alagille syndrome: additional evidence of a vasculopathy. Pediatrics 1999; 103 : 505–508.
29. Martin SR, Garel L, Alvarez F. Alagille´s syndrome associated with cystic renal disease. Arch Dis Child 1996; 74 : 232–235.
30. Berard E, Sarles J, Triolo V, e al. Renovascular hypertension and vascular anomalies in Alagille syndrome. Pediatr Nephrol 1998; 12 : 121–124.
31. Habib R, Dommergues JP, Gubler MC, et al. Glomerular mesangiolipidosis in Alagille syndrome. Pediatr Nephrol 1987; 1 : 455–454.
32. Schonck M, Hoorntje S, van Hooff J. Renal transplantation in Alagille syndrome. Nephrol Dial Transplant 1998; 13 : 197–199.
33. Olsen IE, Ittenbach RF, Rovner AJ, et al. Deficits in size-adjusted bone mass in children with Alagille syndrome. J Pediatr Gastroenterol Nutr 2005; 40 : 76–82.
34. Bucuvalas JC, Horn JA, Carlsson L, et al. Growth hormone insensitivity associated with elevated circulating growth hormone-binding protein in children with Alagille syndrome and short stature. J Clin Endocrinol Metab 1993; 76 : 1477–1482.
35. Kamath BM, Wanrong Y, Heather M, et al. Outcomes of liver transplantation for patients with Alagille syndrome: The studies of pediatric liver transplantation experience, Liver Transplantation 2012; 18 : 940–948.
36. Chong SK, Lindridge J, Moniz C, Mowat AP. Exocrine pancreatic insufficiency in syndromic paucity of interlobular bile ducts. J Pediatr Gastroenterol Nutr 1989; 9 : 445–449.
37. Kamath BM, Kathleen LM, Piccoli DA. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr 2010; 50 : 580–586.
38. Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology 2002; 35 : 1501–1506.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2014 Číslo 4
- Léčba bolesti a horečky u dětí
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- Očkování nejvíc potřebuje ten, kdo sám být očkován nemůže − kazuistika
- Pokrok v boji s malárií − první vakcína poskytující přijatelnou ochranu proti nemoci
Najčítanejšie v tomto čísle
- Alagilleův syndrom
- Sekundární amenorea nebo oligomenorea
- Primární amenorea
- Klinická a laboratorní charakteristika 22 dětí s Kawasakiho nemocí